Chapter 65 Syncope and Paroxysmal Disorders Other than Epilepsy
Paroxysmal disorders, including epilepsy and syncope, represent one of the most common neurological problems in the pediatric population. Although the clinical manifestations of paroxysmal disorders are highly heterogeneous, recent advances in molecular genetic analysis have highlighted striking similarities in their molecular pathophysiology [Crompton and Berkovic, 2009]. Paroxysmal disorders other than epilepsy are described in this chapter (Box 65-1). Breath-holding spells, migraine, pseudoseizures, and sleep-related paroxysmal disorders are reviewed elsewhere in this book.
Box 65-1 Syncope and Other Nonepileptic Paroxysmal Disorders




































Syncope
Syncope is defined as the temporary loss of consciousness and postural tone resulting from transient and diffuse cerebral hypoperfusion, followed by spontaneous recovery with no neurological sequelae [Feit, 1996]. In the young patient, syncope often results from a fall in systolic pressure below 70 mmHg or a mean arterial pressure of 30–40 mmHg [Kaufmann, 2004]. The event is typically preceded by a prodrome lasting several seconds to 1–2 minutes, which has distinctive premonitory features such as nausea, epigastric discomfort, blurred or tunnel vision, muffled hearing, dizziness, lightheadedness, diaphoresis, hyperventilation, palpitations, pallor, cold and clammy skin, or weakness [Sapin, 2004; Strieper, 2005]. These symptoms may occur in any combination or be variably present in any given patient from one episode to the next. If the prodrome is of sufficient duration, patients may learn to recognize their symptoms and lie down to relieve the symptoms and prevent syncope [McLeod, 2003]. Although syncope is commonly a benign self-limiting event, rarely it may be the first warning sign of a serious underlying cardiac or noncardiac disease.
Epidemiology
Syncope is a common clinical problem affecting an estimated 15–25 percent of children and adolescents prior to adulthood. It is not often brought to medical attention, thus accounting for only about 1 percent of pediatric emergency room visits [Vlahos et al., 2007; Longin et al., 2008]. Its true incidence therefore remains unknown. In the 26-year surveillance of the Framingham study, syncope occurred in 3 percent of men and 3–5 percent of women [Savage et al., 1985]. In the Rochester Epidemiologic Project, Driscoll et al. reported on older children and adolescents studied over two 5-year periods [Driscoll et al., 1997]. In the first 5-year period (1950–1954), the incidence of syncope cases requiring medical attention equaled 71.9 per 100,000 population; the second period of study (1987–1991) reported an incidence of 125.8 cases per 100,000 population, with this incidence peaking among adolescents, in particular females aged 15–19 years [Batra and Balaji, 2005]. According to Sheldon et al., the most common age for a child’s first vasovagal syncopal episode is approximately 13 [Sheldon et al., 2006]. The recurrence rate of syncope ranges from 33 to 51 percent when patients are followed for up to 5 years [Kapoor et al., 1983].
Etiology
Syncope may result from cardiovascular or neurological causes (cardiovascular-mediated or neurally mediated syncope). Each of these categories accounts for about 50 percent of adult syncope but, in children, cardiovascular-mediated syncope is less frequent than it is in adults [Kapoor, 2000].
Cardiovascular-Mediated Syncope
Cardiovascular-mediated syncope has a higher mortality and a higher incidence of sudden death than neurally mediated syncope [Kapoor et al., 1983]. It is therefore imperative to distinguish syncope from seizures, and to triage patients with syncope due to malignant cardiac causes for urgent and appropriate investigations and management [Crompton and Berkovic, 2009]. Risk factors that suggest structural or conduction heart defects are given in Box 65-2. If any of these risk factors is present, an urgent referral should be made for pediatric cardiology evaluation. Cardiovascular causes of syncope in children are given in Box 65-3.



















Clinical Features
A diagnosis of syncope rests mainly on clinical grounds [Lerman-Sagie et al., 1994]. The patient’s history, physical examination, and electrocardiography (EKG) have a combined diagnostic yield of about 50 percent [Kaufmann, 2004]. A prodromal phase of presyncope consists of lightheadedness, blurred vision, epigastric discomfort, nausea, pallor, or diaphoresis [Manolis et al., 1990; Feit, 1996; McLeod, 2003]. When present, these clinical features help to differentiate syncope from epilepsy. A detailed history usually reveals contributory environmental factors before the loss of consciousness and postural tone. These environmental factors include upright posture, prolonged standing, change in posture (orthostasis), crowding, heat, fatigue, hunger, or a concurrent illness [Sutton, 1996]. Emotional or stress factors, such as venipuncture, public speaking, “fight-or-flight” situations, pain, and fear, are also commonly identified [Driscoll et al., 1997]. The loss of consciousness is usually brief, lasting from a few seconds to 1–2 minutes, followed by rapid spontaneous recovery without neurological deficits. During the ictus the patient may have tonic posturing or a brief clonic seizure, rarely associated with incontinence. Rarely, myoclonic jerks mimicking an epileptic seizure may occur during syncope [Crompton and Berkovic, 2009]. The postictal period may be accompanied by persistent nausea, pallor, diaphoresis, and a generally “washed-out” appearance. Complete recovery usually evolves in less than an hour [McLeod, 2003].
Distinguishing between neurocardiogenic syncope and seizure is the most common clinical dilemma and a frequent source of diagnostic error. Often, interobserver agreement is poor when a retrospective diagnosis is made on information obtained after a single syncopal episode. In distinguishing patients with syncope due to cardiac causes, a history or physical signs of cardiac disease can be 95 percent sensitive for a cardiac etiology [Crompton and Berkovic, 2009]. These features are, however, nonspecific, as neurocardiogenic syncope might occur in patients with heart disease [Crompton and Berkovic, 2009]. Patients with pseudosyncope and pseudoseizures typically use the events consciously or unconsciously to avoid an unpleasant emotional situation [Wieling and Shen, 2008]. Most of these patients are young females. A very high number of events, episodes without injury, and florid symptomatology are typical. Furthermore, in these patients, recovery after a syncopal event is often prolonged (10–30 minutes), despite a supine posture. In true syncope, consciousness returns within 1 minute of lying down, and unconsciousness for more than 5 minutes is rare [Wieling and Shen, 2008].
The evaluation of syncope may therefore result in unnecessary and costly investigations [Landau and Nelson, 1996]. A complete evaluation would include neuroimaging studies, electroencephalography (EEG), EKG, echocardiography, Holter monitoring, selected metabolic testing, and, in certain cases, intracardiac electrophysiologic studies or implantable continuous loop recordings. Despite extensive investigations, more than 40 percent of patients with recurrent syncope do not have a specific diagnosis [Kapoor, 1991; Grubb et al., 1992].
Pathophysiology
Lewis first introduced the term “vasovagal syncope” in 1932 to indicate that the blood vessels (vaso) and the heart (vagal activity) were involved in the syncopal event [Lewis, 1932]. Hence, the classical clinical signs of vasovagal syncope are marked bradycardia and hypotension. There is no consensus concerning the mechanisms underlying the vasovagal reaction, but several theories have been proposed [Abboud, 1993; Kosinski et al., 1995; Grubb and Kosinski, 1996]. The ventricular theory (Figure 65-1) proposes that, among predisposed individuals who experience recurrent syncope, excess peripheral venous pooling on prolonged standing results in diminished venous return. Decreased cardiac ventricular filling activates mechanoreceptors located mainly in the inferoposterior wall of the left ventricle, which send afferent impulses via C-fibers to the dorsal nucleus of the vagus [van Lieshout et al., 1991]. Arterial baroreceptors and carotid sinus afferent activation may also contribute to the complex pathophysiology of syncope [Kinsella and Tuckey, 2001]. These inhibitory cardiac and arterial receptors mediate increased parasympathetic activity, and inhibit sympathetic activity that results in bradycardia, vasodilatation, and hypotension (Bezold–Jarisch reflex) [Kinsella and Tuckey, 2001; Shen and Gersh, 1993]. The normal response during upright posture is an increased heart rate and diastolic pressure, and an unchanged or slightly decreased systolic pressure [Rea and Thames, 1993]. In individuals susceptible to recurrent syncope, a “paradoxical” reflex bradycardia and peripheral vascular dilatation occur [Grubb and Kosinski, 1996]. The previous emphasis on parasympathetic (vagal) output is shifting to sympathetic withdrawal as the main mechanism responsible for the bradycardia or asystole (cardioinhibitory response) and hypotension (vasodepressor response) that accompany neurocardiogenic syncope [Hannon and Knilans, 1993]. Although parasympathetic-mediated bradycardia remains a contributory factor in syncope, the responsible phenomena are vasodilatation and hypotension [Kosinski et al., 1995]. Current hypotheses propose that the primary efferent event is systemic vasodilatation, and that this vasodepressor element is mediated by a profound, centrally mediated sympathetic withdrawal [Kosinski et al., 1995]. The persistence of neurocardiogenic syncope in subjects who had cardiac transplants and, therefore, technically denervated hearts, as well as the findings of increased epinephrine levels during upright position and syncope, are strong evidence supporting a sympathetic withdrawal mechanism in syncope [Fitzpatrick et al., 1993; Njemanze, 1993; Sra et al., 1994]. Altered cerebral autoregulation may also be contributory in neurocardiogenic syncope [Rodriguez-Nunez et al., 1997]. Transcranial Doppler studies have demonstrated cerebral vasoconstriction during tilt-table-induced syncope [Grubb et al., 1991]. These observations may stimulate future research and modify current insight about neurocardiogenic syncope.
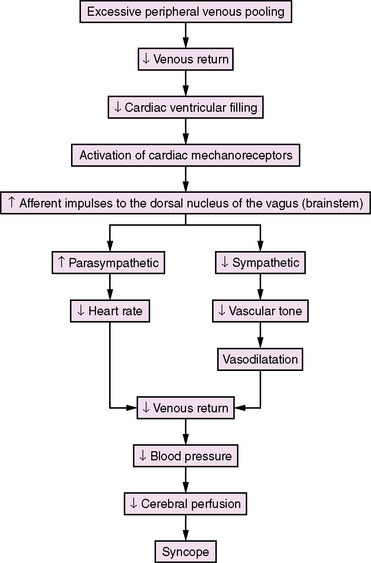
Diagnostic Evaluation
The history, clinical examination, and EKG have a combined diagnostic yield of 50 percent [Kaufmann, 2004]. The patient history is the cornerstone on which the diagnosis of syncope is made. Important historical details to take from patients are given in Box 65-4. On clinical examination, blood pressure and heart rate should be taken in the supine and upright positions, noting any orthostatic hypotension with or without an increase in heart rate. Special attention should be paid to detecting cardiac anomalies. An EKG should be obtained on all patients who present with syncope, especially if it is recurrent or occurs with exercise. All patients with recurrent syncope, family history of syncope or sudden unexplained death should be referred to cardiology for further evaluation. This may include echocardiography and a Holter or event monitor. Rarely, cardiac catheterization with right ventricular endomyocardial biopsy may be necessary before a patient can resume activities [Strieper, 2005].











Tilt-Table Testing
Until the mid-1980s, the diagnosis of neurocardiogenic syncope was made primarily by a careful and detailed patient and family history, the physical examination, and EKG. Tilt-table testing as a potential diagnostic tool for neurocardiogenic syncope was introduced only in 1986 after the ground-breaking report by Kenny et al. [Kenny et al., 1986]. A number of reports have since emerged, attesting to the utility of the test in reproducing syncopal episodes in patients who are predisposed to neurocardiogenic hypotension and bradycardia.
Despite criticism that this provocative test suffers from “naïve rationale,” lacks patient selection standards, and needs controlled treatment and outcome studies [Landau and Nelson, 1996], the tilt-table test continues to enjoy popularity as a noninvasive and physiologically appropriate neurophysiologic test for the diagnosis of neurocardiogenic syncope [Kapoor, 1999; Dijane et al., 1996].
The test is done by positioning the patient head upright at an angle of 60–80 degrees for 15–60 minutes on a tilt table with a supporting footboard. A tilt-table test result is positive when the symptoms of syncope or presyncope are reproduced [Sra et al., 1991]. If the result is negative, isoproterenol is administered intravenously and the dose increased until the heart rate goes up by at least 20 percent. To standardize the test, the duration of head-up tilting has been increased to 45 minutes or 2 standard deviations (SD) of the mean time required to reproduce syncope [Sneddon and Camm, 1993]. A comprehensive analysis of available tilt-table data suggests that administration of isoproterenol has no added benefit, and 60 degrees for 45–60 minutes is recommended [Kapoor et al., 1994].
Experience with tilt-table test among pediatric populations is limited. Of 35 adolescents who had recurrent presyncope or syncope, 26 had positive tilt-table test results at 60 degrees for 30–60 minutes and isoproterenol infusion from 1 to 5 μg/minute [Thilenius et al., 1991]. Among 54 pediatric patients who had recurrent syncope, tilt-table testing at 80 degrees for 30 minutes was superior to studies, such as chest radiograph, EKG, echocardiogram, EEG, or neuroimaging, in arriving at a diagnosis of neurocardiogenic syncope [Strieper et al., 1994]. Tilt-table studies in 20 children with unexplained syncope and 10 controls, using 60 degrees for 25 minutes and isoproterenol infusion of 0.02–0.08 μg/kg/minute, were positive in 75 percent of patients and in 10 percent of controls, with a sensitivity of 75 percent and a specificity of 90 percent [Alehan et al., 1996]. The tilt-table test has been used in children as young as 3 years of age [Grubb et al., 1992].
In addition to heart rate and blood pressure monitoring during tilt-table testing, EKG recording may be used to differentiate anoxic from epileptic seizures [Grubb et al., 1992]. More controlled studies and standardization of degree and duration of tilting are necessary to validate the tilt-table test as a safe, practical, and useful diagnostic tool for neurocardiogenic syncope in children [Benditt and Lurie, 1996; Mansourati and Blanc, 1996; Moya et al., 1996a, b; Victor, 1996].
Treatment
The objective of treatment for neurocardiogenic syncope is to prevent recurrent syncope, which leads to impaired quality of life, psychological distress, and substantial morbidity. Once a diagnosis of neurocardiogenic syncope is confirmed, treatment requires counseling of the patient (when appropriate) and his or her parents. The benign nature of these events should be explained to allay concerns about epilepsy or sudden death. Neurocardiogenic syncope almost always resolves within months to 3–5 years after onset [Strieper, 2005]. Most patients presenting after a single uncomplicated syncopal event require simple reassurance, education about the disease, and advice on recognizing prodromal symptoms and how to avoid provocative situations – in particular, prolonged standing, sudden postural changes, dehydration, and irregular meal times. If a prodromal phase is consistently present, the patient may by taught to recline or sit to avoid injury from a fall. Supplemental fluids and electrolytes may be beneficial; up to 1500–2500 mL per day are recommended for adolescents. Patients should be instructed to increase dietary salt, either as salt tablets or liberal use of salt with meals (Box 65-5).













If, despite these conservative measures, the syncopal episodes become refractory, pharmacologic therapy may be tried. Favorable but not consistent response to treatment has been reported with β-adrenergic receptor antagonists, α-adrenergic receptor agonists, anticholinergic agents, theophylline, serotonin reuptake inhibitors, and mineralocorticosteroids [Milstein et al., 1990; Scott et al., 1995; Raviele et al., 1996; Boehm et al., 1997; Sra et al., 1997] (Box 65-6). Beta blockers, fludrocortisone and midodrine, and an α-adrenergic agonist, are often prescribed in children but none of these agents has shown a consistent therapeutic benefit in clinical trials [Kaufmann and Freeman, 2004; Freeman, 2008]. Low-dose midodrine is promising and is currently recommended as first-line therapy for vasovagal syncope in children by some authorities [Stewart, 2006]. When using fludrocortisone, combine with increased salt intake for an optimal effect. Subjects who require isoproterenol to induce syncope during tilt-table testing or who experience tachycardia before syncope may respond better to beta-blocker therapy [Sra et al., 1992; Leor et al., 1994; Wieling and Shen, 2008].
Box 65-6 Medications for Treatment of Neurocardiogenic Syncope*
















* Pediatric doses are given for those medications used in children for which dosages have been recommended.
(Data from Grubb and Kosinski, 1996; Lazarus and Mauro, 1996; Raviele et al., 1996; Sra et al., 1997.)
Prognosis
The prognosis for recovery is excellent in neurocardiogenic syncope. Most patients show spontaneous resolution of their syncope and presyncope within the first year after onset; 5–10 percent will however, continue to show symptoms over an extended period of time, often up to 5 years [Strieper, 2005].
Convulsive Syncope
A brief tonic or, rarely, a clonic seizure may accompany syncope. In a study of blood donors, 0.05 percent suffered a convulsion associated with syncope (convulsive syncope) [Lin et al., 1982]. Among 216 children who had a positive tilt-table test, 25 (11.6 percent) had seizures during the test [Fernandez Sanmartin et al., 2003]. Most convulsions consisted of tonic spasms (65 percent), characterized by eye rolling, nuchal rigidity, arms flexed at the elbow, and fists clenched, followed usually by prompt recovery [Lin et al., 1982]. Other types of convulsions were myoclonic (23 percent), clonic (6 percent), and tonic-clonic (6 percent). No difference was found in the severity of bradycardia or hypotension among those who had syncope associated with convulsion and those who did not [Lin et al., 1982]. In subjects who had cardiac asystole induced by ocular compression, only those who remained asystolic for more than 14 seconds experienced convulsive phenomena [Lin et al., 1982]. Convulsive syncope results from cerebral ischemia and is not indicative of an epileptic predisposition. The EEG reveals diffuse slowing, followed by loss of electrocerebral activity; epileptiform activity is absent [Fernandez Sanmartin et al., 2003; Stephenson, 1990d]. Rarely, an epileptic seizure is triggered by syncope. In such cases, the EEG, in contrast to what is seen in convulsive syncope, reveals epileptiform activity [Stephenson, 1990g].
Reflex Syncope
Syncope that is triggered by specific factors or events is known as reflex or situational syncope. The most common of these among infants and children is breath-holding spells. These are discussed in greater detail in Chapter 64. A related but distinctive type of syncope (known by various names in the past, including pallid breath-holding spells or pallid infantile syncope) is frequently confused with breath-holding spells. A simple and more appropriate designation for this type of paroxysm is reflex syncope.
In reflex syncope, the antecedent event is minor trauma (usually to the head) before loss of postural tone and consciousness, without any audible inspiratory stridor or expiratory cry. Reflex syncopal episodes are easily confused with typical breath-holding spells, but evidence is lacking that these result from transient cerebral hypoxia because of breath holding. The rapid or immediate onset of syncope after minor trauma or other unexpected painful stimuli differentiates reflex syncope from breath-holding spells. In breath-holding spells, several seconds may elapse before loss of consciousness. There is general agreement that reflex syncopal attacks do not represent epileptic seizures, but the mechanism responsible is less clear. Some refer to these events as reflex anoxic seizures [Stephenson, 1978]. According to Stephenson, cardiac arrest is inducible in individuals susceptible to reflex syncope by the ocular compression test. This test is performed by applying pressure over the closed eyelids for 10 seconds during cardiac and EEG monitoring [Stephenson, 1980]. After 7–15 seconds of asystole, a typical paroxysm is reproduced, thus confirming the diagnosis of reflex syncope. Reproduction of reflex syncope by ocular compression suggests that these children have an exaggerated oculocardiac reflex [Stephenson, 1990f]. Despite assurances about the safety and diagnostic usefulness of the ocular compression test to reproduce this type of syncope [Gastaut, 1974; Stephenson, 1980, 1990f], its acceptance as a provocative test has been limited.
Situational Syncope
Other triggering factors associated with syncope include cough, deglutition (cold liquids), defecation, diving, micturition, sneezing, trumpet playing, weight lifting, and Valsalva maneuver [Hannon and Knilans, 1993]. These events are more common in adults than in children and are referred to as situational syncope. A common denominator in situational syncope is the fact that most of the triggering factors are accompanied by a Valsalva-like maneuver. Hair-grooming syncope is an uncommon type of situational syncope among adolescent females; it is often followed by brief seizure activity [Lewis and Howell, 1986; Igarashi et al., 1988; Lewis and Frank, 1993]. The convulsive syncope is almost invariably preceded by a prodrome of presyncope symptoms of nausea, lightheadedness, diaphoresis, and visual blurring. The reaction is thought to be a variant of neurocardiogenic syncope that is triggered by hair pulling or scalp stimulation, which activates the trigeminal nerve [Kosinski and Grubb, 2005].
Suffocation or Strangulation Syncope
Meadow’s syndrome (Munchausen’s by proxy) is a rarely suspected cause of syncope. A caretaker induces loss of consciousness by obstructing the infant’s airway using a pillow or by pressing the infant’s face against the caretaker’s trunk [Stephenson, 1990e]. In other cases, compression of the neck by strangulation results in cerebral hypoxia and, after repeated attempts, brain damage. A solicitous and omnipresent caretaker should raise suspicion of foul play [Folks, 1995]. The morbid events have been documented in some cases during video/EEG monitoring, but the incriminating evidence may not be admissible in court.
Drug-Induced Syncope
When episodes begin with a slow onset and gradual recovery, consider a toxic or metabolic cause, such as hypoglycemia, alcohol, or drugs (illicit or prescribed) [Olshansky, 2005]. Among the medications that can cause syncope are those that induce ventricular tachycardia or cause hypotension. Cardiovascular medications (vasodilators and antiarrhythmics), psychotropics, diuretics, and glucose-controlling medications are the most common drugs associated with syncope [Lazarus and Mauro, 1996]. Illicit drugs may also cause syncope (particularly alcohol, but also cocaine and marijuana). Alcohol and illicit drugs can cause syncope by several mechanisms, including exacerbation of a supraventricular or ventricular tachyarrhythmia [Strieper, 2005]. Drug-induced syncope is common among the elderly but rare among children. However, illicit drug use is increasing significantly in the younger age group. Toxicology screens often provide clues to this diagnosis.
Psychogenic Syncope
One of the most important causes of syncope in pediatric patients, in particular adolescents, is psychogenic syncope. Several features help distinguish psychogenic syncope from neurocardiogenic syncope [Benbadis and Chichkova, 2006; Thijs et al., 2009]:
Paroxysmal Dyskinesias
Paroxysmal dyskinesias are episodic attacks of involuntary hyperkinetic (dystonic, choreoathetoid, or ballistic) movements with preserved consciousness. In 1940, Mount and Reback first described a familial paroxysmal movement disorder manifested by involuntary writhing and posturing of the trunk and extremities, later labeled as paroxysmal dystonic choreoathetosis [Mount and Reback, 1940]. Kertesz introduced the term paroxysmal kinesigenic choreoathetosis to differentiate cases in which the episodes were brief and precipitated by movement, instead of by prolonged immobility or intake of specific beverages (coffee, tea, cola drinks, alcohol), as in the cases of Mount and Reback [Kertesz, 1967]. These hyperkinetic movements or dyskinesias may manifest as dystonia (abnormal distorted posturing), chorea (arrhythmic, bizarre, jerky, dancing movement), athetosis (distal slow, sinuous limb movement), or ballism (violent flailing limb movement). They may occur individually or in various combinations. The most widely used classification system for the paroxysmal dyskinesias is the one proposed by Demirkiran and Jankovic in1995 [Demirkiran and Jankovic, 1995]. The paroxysmal dyskinesias are classified into four types: paroxysmal kinesigenic dyskinesia (PKD), paroxysmal nonkinesigenic dyskinesia (PNKD), paroxysmal exercise-induced dyskinesia (PED), and paroxysmal hypnogenic dyskinesia (PHD). Each category is further subdivided into idiopathic (familial and sporadic) and, rarely, symptomatic [Blakeley and Jankovic, 2002] secondary to stroke, trauma, multiple sclerosis, central nervous system infections, and other known causes, with resultant abnormality on brain magnetic resonance imaging. The majority of cases are primary or idiopathic, and of the PKD or PNKD type. PHD, though included in this classification scheme due to semiologic similarity, has been proven to consist of nocturnal seizures secondary to autosomal-dominant nocturnal frontal lobe epilepsy (ADNFLE) [Luders, 1996; Phillips et al., 2001].
The etiology, anatomy, physiology, and pathogenesis of the paroxysmal dyskinesias are not yet completely understood. Evidence supports the striatum as the primary area of brain involvement, based on current clinical, radiological, and experimental data. Symptomatic cases have been shown to have structural pathology of the striatum; abnormalities of the striatum have been found on MR spectroscopy, positron emission tomography (PET), and single photon emission computerized tomography (SPECT); and in in vivo experimental studies, using the dtsz mutant hamster animal model of paroxysmal dyskinesia, striatal increase of extracellular dopamine during dystonic episodes has been shown [Ko et al., 2001; Sanger, 2003; Hamann and Richter, 2004]. However, in another animal study using the lethargic mouse model (produced by calcium channel mutation), abnormal cerebellar output was shown to be responsible for the paroxysmal dyskinesia [Devanagondi et al., 2007]. Excess cytochrome oxidase activity in the red nucleus disappeared after surgical removal of the cerebellum. A similar conclusion was reached in the tottering mouse model [Campbell et al., 1999].
The etiology of most of these paroxysmal dyskinesias, as with many periodic syndromes, is thought to be secondary to an underlying channelopathy. They share common clinical characteristics with epilepsy, migraine headaches, periodic paralysis, episodic ataxia with myokymia, including episodic attacks with normal interictal examination, common precipitating mechanisms such as stress, fatigue, diet, alcohol, and caffeine, and treatment response to similar medications. Both hypokalemic and hyperkalemic periodic paralysis and episodic ataxia with myokymia are due to mutations in ion-channel genes. Carbamazepine is effective in certain epilepsies, as in PKD, and acetazolamide is useful in periodic paralysis, myotonia, episodic ataxia, and some paroxysmal dyskinesias [Bhatia et al., 2000; Ptacek and Fu, 2001; Celesia, 2001; Margari et al., 2005; Cannon, 2001]. Genetic mutations in the alpha subunit of calcium-sensitive potassium channel on chromosome 10q22 cause epilepsy and PNKD [Du et al., 2005]. Mutations in voltage-gated neuronal K+ channel Kv7 (KCNQ) produces dyskinesia in the dtsz mutant hamster model of paroxysmal nonkinesigenic dyskinesia. Retigabine and flupirtine, which result in opening of the above type of K+ channel, effectively treated the paroxysmal dystonia, proving the role of Kv7 channel mutation in the causation of PNKD [Richter et al., 2006]. Interestingly, however, many of the familial PNKDs are caused by the mutation of MR-1 (myofibrillogenesis regulator 1) gene on chromosome 2. The MR-1 gene is not involved with an ion channel; rather, it is predicted to be involved in the stress response pathway [Lee et al., 2004; Rainier et al., 2004; Chen et al., 2005a; Hempelmann et al., 2006; Bruno et al., 2007]. Some symptomatic forms of PKDs are caused by the X-linked monocarboxylate transporter 8 (MCT 8) gene mutation responsible for transport of T3 (tri-iodothyronin) across the neuronal membrane [Dumitrescu et al., 2004; Brockmann et al., 2005; Fuchs et al., 2009].
The paroxysmal dyskinesias are also described in Chapter 68.
Paroxysmal Kinesigenic Dyskinesia
This is the most common form of all the paroxysmal dyskinesias, although the exact prevalence is unknown. The attacks are brief (usually lasting less than 1 minute) and characterized by dystonic posturing, choreoathetosis, or ballistic movements, either singly or in combination; they affect one or both sides concomitantly [Lance, 1977; Bruno et al., 2004]. The patient remains conscious throughout the episode, without any postictal impairment. In the largest series of 121 cases of paroxysmal kinesigenic dyskinesia, 51 were male and 44 female [Bruno et al., 2004]. Ninety-five cases were familial, and the majority of these were consistent with autosomal-dominant inheritance. Males also outnumbered females in sporadic cases. The mean age at onset was 11.7 ± 3.4 years in the familial cases, and similar for the sporadic ones. The age range, however, varied from 1 to 20 years. The dyskinesias are always triggered by sudden movement; however, an intention to move or startle may also bring them on. Anxiety or stress may also play an important role, causing confusion, at times, with a psychogenic movement disorder with consequent delay in the diagnosis. Spells occur more frequently following physical exertion or during sickness. An aura is present in more than 80 percent of cases. These auras consist of a feeling of tightness, numbness, tingling, or paresthesia in the involved extremities [Lance, 1977; Bruno et al., 2004]. The ictus consists of dystonic posturing in up to two-thirds of all cases. Choreoathetosis or ballistic movements occur in some, and a combination of movements occurs in one-third. In about one-third of cases, the attacks may be focal or unilateral, one-third may have bilateral involvement, and the remainder has either unilateral or alternating bilateral involvement. In focal cases, the movement usually starts with the limb in action. The attacks are typically short, with more than 90 percent lasting less than 30 seconds, and usually never more than a minute. The attacks occur daily in more than 80 percent of cases, and the frequency may reach up to 100 attacks in a day [Lance, 1977; Houser et al., 1999; Bruno et al., 2004].
Associated neurological disorders, like infantile convulsions, are common (11–19 percent) in children with PKD; almost half of their family members had infantile seizure [Margari et al., 2000, 2002; Bhatia, 2001; Bruno, et al., 2004]. This association brought up a homogenous autosomal-dominant syndrome described as infantile convulsions and choreoathetosis (ICCA), with benign seizures in infancy, followed by later-onset paroxysmal kinesigenic dyskinesia [Rochette et al., 2008]. In familial forms of PKD, febrile seizures are seen in 9 percent of the affected individuals and in 30 percent of other family members. Migraine with and without aura occurs in about one-third of all PKD cases and in two-thirds of their family members. Other neurological abnormalities include writer’s cramp, essential tremor, and myoclonus [Bruno et al., 2004; Margari et al., 2005].
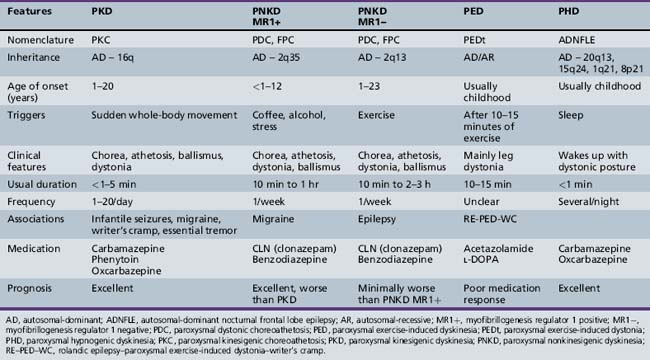
Differential diagnoses depend on age of onset of the symptoms. These include other forms of paroxysmal dyskinesias, dopa-responsive dystonia, complex motor tics, complex motor stereotypies, seizure, pseudoseizure, psychogenic movement disorder, malingering, shuddering attacks, and Sydenham’s chorea. Complex motor stereotypies in younger children are sometimes difficult to distinguish from PKD or PNKD. Excitement precipitating the complex movement pattern and involvement of predominantly the upper limbs or upper part of the body in a child with autistic behaviors or cognitive impairment is suggestive of a complex motor stereotypy rather than PKD. Dopa-responsive dystonia can be distinguished clinically by the predominant involvement of the lower limbs and a diurnal variation, with worsening in the evening. Chorea in Sydenham’s chorea may start unilaterally and intermittently on attempted activity, but the preceding history of sore throat, laboratory evidence of streptococcal infection, and self-limiting course of the disorder help in making the diagnosis. The distinguishing features of various types of paroxysmal dyskinesias are shown in Table 65-1. Increased awareness of PKD among clinicians is needed, as the diagnosis is delayed in most cases. The mean time from first seeking medical attention to confirmation of diagnosis is approximately 5 years [Bruno et al., 2004]. Proposed diagnostic criteria for PKD are shown in Box 65-7 [Bruno et al., 2004].






Despite the existence of several PKD gene loci found through linkage studies, no PKD gene has been identified so far. A form of autosomal-dominant PKD with episodic ataxia and spasticity has been mapped at chromosome 1p [Auburger et al., 1996]. ICCA, or ICCA-like syndromes, has been mapped to the pericentromeric region of chromosome 16 (16p12–q12). This involves the PKC1 or EKD1 gene locus [Tomita et al., 1999; Swoboda et al., 2000; Bennett et al., 2000; Rochette et al., 2008]. Another locus (EKD 2) has been identified on the same chromosome in an Indian family with PKD [Valente et al., 2000].
Neurophysiology and neuroimaging studies are normal in familial and sporadic idiopathic cases, except for occasional slowing noted on the EEG [Buruma and Roos, 1986]. No epileptiform activity is usually seen, even during an episode. Nevertheless, reports of epilepsy among members of the same family raise the question about the possible association between these disorders [Demirkiran and Jankovic, 1995; Margari et al., 2000, 2002; Rochette et al., 2008]. Lombroso demonstrated ictal discharges emanating from the supplementary sensorimotor cortex and ipsilateral caudate nucleus by invasive long-term monitoring in a female with paroxysmal kinesigenic choreoathetosis [Lombroso, 1995]. Thus, some authors believe that these paroxysmal movement disorders are an expression of subcortical seizures involving cortico-striato-thalamo-cortical circuits [Tan et al., 1998]. The pathology in this circuit underlies the pathophysiology of most movement disorders in general. In primary (idiopathic or genetic) movement disorders, there is no detectable pathology on MRI of the brain, whereas in secondary or symptomatic cases there is involvement of basal ganglia or thalamus, with or without cortical involvement. Electromyography shows doublet or triplet discharges. Somatosensory-evoked potential (SSEP) shows reduction in amplitude of cortical sensory response. Motor-evoked potential, however, shows an increased motor cortical excitability with diminished stimulation threshold, high amplitude, and motor facilitation. Spinal and brainstem reflexes, like H reflex and blink reflex, suggest lack of inhibition. This phenomenon has led to the alternate hypothesis of spinal or brainstem modulation of central motor activity, producing paroxysmal dyskinesias [Lee et al., 1999].
Once diagnosed, there are only a few neurological disorders with a more satisfying treatment response, with an exquisite sensitivity to antiepileptic medications [Wein et al., 1996]. The attacks can be prevented, even with subtherapeutic levels of phenytoin or carbamazepine. Other medications found to be effective, but to a lesser degree, are barbiturates, benzodiazepines, valproate, lamotrigine, levetiracetam, scopolamine, l-DOPA, belladonna, chlordiazepoxide, and diphenhydramine [Lance, 1977; Kinast et al., 1980; Bruno et al., 2004]. Most children tried with other medication were eventually switched back to either phenytoin or carbamazepine. In a recent report, oxcarbazepine worked very well in four children with PKD [Chillag and Deroos, 2009]. This marked sensitivity to antiepileptic medications has been used to differentiate paroxysmal kinesigenic dyskinesia from the nonkinesigenic type. Whether treated or not, the attack frequency generally decreases with increasing age. More than one-quarter of patients enjoy complete remission by age 20 years, and a further quarter have marked reduction in attack frequency. Females in the idiopathic category have the best prognosis [Bruno et al., 2004].
Paroxysmal Nonkinesigenic Dyskinesia
PNKD is characterized by episodes of dystonia and/or choreoathetosis involving the face, trunk, and extremities, often associated with dysarthria and dysphasia, lasting from minutes to several hours and occurring up to several times a week [Pryles et al., 1952; Lance, 1977; Tibbles and Barnes, 1980; Kinast et al., 1980; Bressman et al., 1988; Demirkiran and Jankovic, 1995]. Historically, in 1940, Mount and Reback reported the first family with PNKD, labeling them as cases of “familial paroxysmal choreoathetosis” [Mount and Reback, 1940]. Various eponyms have been used to describe this condition but, due to overlapping semiologic features, the term paroxysmal nonkinesigenic dyskinesia is preferred. Proposed diagnostic criteria for PNKD are indicated in Box 65-8.
 Hyperkinetic involuntary movement attacks, with dystonia, chorea, or combination of these, typically lasting 10 minutes to 1 hour, but up to 4 hours
Hyperkinetic involuntary movement attacks, with dystonia, chorea, or combination of these, typically lasting 10 minutes to 1 hour, but up to 4 hours



Mutations of the myofibrillogenesis regulator 1 (MR-1) gene (also known as the PNKD1 gene) form a distinct homogenous subset of PNKD [Bruno et al., 2007]. Onset is usually in infancy or early childhood, with a mean of 4 years, but late onset has been reported. This is in contradistinction to the MR-1 gene-negative group, which has a later age of onset with a mean of 12 ± 10.8 years. The attacks begin spontaneously at rest or after intake of caffeine or alcohol. Alcohol and caffeine sensitivity is noted in almost 100 percent of MR-1 gene-positive cases, which is distinctly uncommon in the other group. Stress is another common precipitating factor that occurs in more than 80 percent of the MR-1 gene-positive group. Other precipitating factors include fatigue, hunger, chocolate, and excitement. Exercise has been noted to bring on the majority of the attacks in MR-1 gene-negative cases, implicating more heterogeneity in these patients and causing confusion with some cases of paroxysmal exercise-induced dyskinesia. An aura, consisting of focal limb stiffening, twitching, numbness, a generalized “funny feeling,” or lightheadedness, has been noted [Williams and Stevens, 1963; Bruno et al., 2007]. About 12 percent present only with dystonia, and the rest with a combination of dystonia and chorea in the MR-1 gene-positive cases, whereas the other group may have dystonia, chorea, a combination of both, and ballism as part of the clinical manifestations. Almost half may have speech involvement. Typical attack duration is 10 minutes to 1 hour, but may last up to 12 hours. Attack frequency is highly variable, but 86 percent have at least one attack every week.
Clonazepam and diazepam are the most effective preventive and abortive agents in 97 percent of cases studied by Bruno et al. [Bruno et al., 2007]. Sleep benefit is also noted in the majority of cases [Byrne et al., 1991; Bruno et al., 2007]. The MR-1-negative group, however, did not have sustained benefit from clonazepam in at least half of the cases treated. Other antiepileptic medications, including valproate, carbamazepine, topiramate, and gabapentin, proved partially beneficial for MR-1-negative patients, but MR-1-positive patients did not respond well. l-DOPA, acetazolamide, and haloperidol did not help either group.
Prognosis is not as good as in PKD; however, almost two-thirds of the mutation-positive group improved with age, with only a minority (18 percent) becoming worse. The prognosis in the mutation-negative group is a little worse than in the mutation-positive group. Migraine is coexistent in 47 percent of cases in the MR-1-positive cases, whereas seizures are reported in 23 percent of the MR-1-negative cases [Kinast et al., 1980; Przuntek and Monninger, 1983; Bruno et al., 2007].
PNKD follows an autosomal-dominant transmission pattern, with almost equal sex distribution. The majority of familial PNKD are caused by the mutation of the MR-1 gene on chromosome 2q35 [Fouad et al., 1996; Fink et al., 1996; Jarman et al., 1997; Raskind et al., 1998]. Recently, linkage studies in a Canadian family with European descent identified a second locus at the 2q31 region [Spacey et al., 2006]. The MR-1 gene is not involved in the ion channel, contrary to previous postulations; rather, it works in the stress response pathway. Although the exact function of MR-1 is unknown, its gene product is homologous to the hydroxyacylglutathione hydrolase (HAGH) of the glyoxalase system. HAGH catalyzes the final step in conversion of methylglyoxal (a byproduct of glycolysis, also found in coffee and alcoholic beverages) to lactic acid and reduced glutathione. This relationship explains the exquisite sensitivity to coffee and alcohol in attack precipitation; emotional stress may also act in a similar manner [Lee et al., 2004; Rainier et al., 2004; Chen et al. 2005a; Hempelmann et al., 2006; Bruno et al., 2007]. Mutations in voltage-gated neuronal K+ channel Kv7 (KCNQ) has been shown to produce dyskinesia in dtsz mutant hamster model of PNKD [Richter et al., 2006]. In some patients with coexistent generalized epilepsy and PNKD, a mutation in the alpha subunit of the calcium-sensitive potassium channel (BK channel) located on chromosome 10q22 has been discovered [Du et al., 2005].
Paroxysmal Exercise-Induced Dyskinesia
This interesting autosomal-dominant disorder is not as common or well understood as the others. No specific genetic defect has yet been found. Children or young adults with otherwise normal health develop dystonic posturing, with or without pain, after a prolonged period of exercise, usually more than 10–15 minutes. Fasting and stress can bring on the attacks. The attack may last 10–30 minutes and usually involves the limb/s being used in exercise. Association with absence or complex partial seizures has been described [Plant et al., 1984; Munchau et al., 2000]. In a Sardinian family with rolandic epilepsy, paroxysmal exertional dyskinesia, and writer’s cramp, and showing an autosomal-recessive pattern of inheritance, the disorder has been has been mapped to 16p12–11.2, which overlaps the ICCA locus [Guerrini et al., 1999]. Linkage of one family pedigree with typical PED was not established to the PNKD or ICCA loci [Munchau et al., 2000]. PED cases may represent a variant of either PKD or PNKD where there is precipitation by exercise. Cases of dopa-responsive dystonias may pose a great challenge in differential diagnosis when the lower limbs are primarily involved; however, the excellent response to l-DOPA in dopa-responsive dystonias, along with gene testing, should clarify that doubt. The responses to medication in PED are modest at best; drugs used have included acetazolamide, l-DOPA, and trihexyphenidyl.
Dopa-Responsive Dystonia
Only a few disorders in the practice of pediatric neurology provide the opportunity to reverse a disabling condition in the dramatic manner afforded by dopa-responsive dystonia (DRD). The condition was first reported by Segawa in 1971, with an autosomal-dominant mode of inheritance [Segawa et al., 1986]. The estimated prevalence in England and Japan is 0.5 per million [Nygaard, 1993]. The onset is between 1 and 9 years of age (average 5 years). Females outnumber males by 3–4:1. Symptoms usually start with unilateral leg or foot dystonia, precipitated by actions such as walking or running, and improvement with rest. The most characteristic feature is the diurnal variation of the dystonia, which worsens in the evening and improves markedly in the morning. Sleep acts as the most important relieving factor. Classically, the dystonia starts with one foot, spreading to affect the whole of the ipsilateral lower limb. It then spreads to the ipsilateral upper limb, contralateral lower limb, and, finally, the contralateral upper limb, following the pattern of the letter N. The last area involved is the craniocervical region. Involvement spreading from one lower limb to the other and, finally, involving upper limbs and craniocervical region may occur. It may take 5–6 years from the onset for the dystonia to be generalized [Segawa et al., 1986; Trender-Gerhard et al., 2009]. The periodicity, action induction, fatigability, asymmetry, and diurnal variation may not be very prominent with advancing disease pathology. This is an important point to consider in the history. Dystonic posturing of the big toe (striatal toe) may mimic an upgoing plantar (Babinski) response [Iivanainen and Kaakkola, 1993]. Features of parkinsonism, such as rigidity, bradykinesia, postural instability, action or postural tremor of the arms, and, rarely, a resting tremor, may eventually develop in some cases [Nygaard et al., 1994]. The presence of rigidity and dystonia in the lower limbs may mimic a spastic diplegic form of cerebral palsy, resulting in misdiagnosis. If in doubt, a trial of l-DOPA usually results in dramatic improvement. It is worth noting that the improvement is expected, even if this condition is diagnosed very late. There is a need for a high index of suspicion on the part of the clinician to think about this disease in the clinical context and consider a trial of l-DOPA [Nygaard et al., 1994; Segawa, 2000; Segawa et al., 2003]. Less frequently, DRD may be inherited in an autosomal-recessive fashion with much younger age of onset, but retaining all the other characteristics of the above description. Dystonia as the predominant clinical entity represents the “pure form” of DRD. In a subset of children, often referred to as having DRD plus syndrome, seizures, developmental delay, other dyskinesias, tremor, chorea, and oculogyric phenomena may occur [Schiller et al., 2004; Clot et al., 2009].
Neuroimaging studies are typically normal. PET studies have been normal in most cases [Snow et al., 1993]. Pathologically, there is no cell degeneration in the nigrostriatal dopaminergic system. There is inadequate dopamine synthesis in the striatum secondary to dysfunction of the enzyme tyrosine hydroxylase, either as a primary deficiency or secondary to a lack of the co-factor tetrahydrobiopterin (BH4). There is reduction of the dopamine metabolite homovanillic acid (HVA) in the cerebrospinal fluid. BH4 acts as essential co-factor for tyrosine, phenylalanine, and tryptophan hydroxylases. Defect in the biosynthetic pathway of BH4 causes a reduction of dopamine and serotonin (5-hydroxytryptamine) metabolites, HVA, and 5-hydroxy indole acetic acid (5-HIAA), respectively [Nygaard, 1995]. Neuropathologic data in one case demonstrated marked reduction of dopamine levels in the substantia nigra and in the striatum [Rajput et al., 1994].
Autosomal-dominant DRD is caused by mutations in the GCH1 gene on chromosome 14 (14q22.1–q22.2), which encodes the enzyme guanosine triphosphate cyclohydrolase 1 (GTPCH-1) in the BH4 biosynthetic pathway [Furukawa et al., 1995; Tanaka et al., 1995; Segawa, 2000; Segawa et al., 2003; Clot et al., 2009]. Autosomal-recessive DRD may be secondary to tyrosine hydroxylase (TH) gene mutation. Tyrosine hydroxylase deficiency is associated with a broader phenotype than just a dopa-responsive dystonia and gait disorder, and includes a more severe infantile parkinsonism or progressive infantile encephalopathy phenotype. DRD plus disorders may be due to sepiapterin reductase gene mutation or other defects in the BH4 biosynthesis pathway.
The response to treatment with l-DOPA in typical cases of DRD is dramatic. Fatigability is usually alleviated within a week, and dystonia within 6 weeks of optimum dose. A combination of l-DOPA and dopa decarboxylase inhibitor should be used. Preparations with a higher ratio of dopa decarboxylase inhibitor, e.g., carbidopa-l-DOPA 25–100 mg, are generally preferred to the 10–100 mg preparation. Young adolescents, more particularly females, have much higher dopa decarboxylase activity, thus requiring a higher dose of carbidopa. A dose of 5 mg/kg/day of carbidopa-l-DOPA is effective in most cases. The medication is generally well tolerated and the effect sustained, even after prolonged use of 30 years [Segawa et al., 1986]. The reason for the lack of any significant side effect is the paucity of dopamine synthesis in this condition with preserved structure of the nigrostriatal system with no cell damage or death. The use of l-DOPA is, therefore, the equivalent of supplementing dopamine in the deficient state.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


