CHAPTER 183 Syndromic Craniosynostosis
Description and Classification of the Faciocraniosynsotoses
Crouzon’s Syndrome
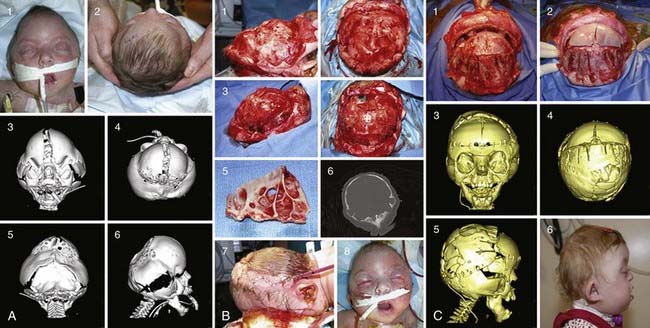
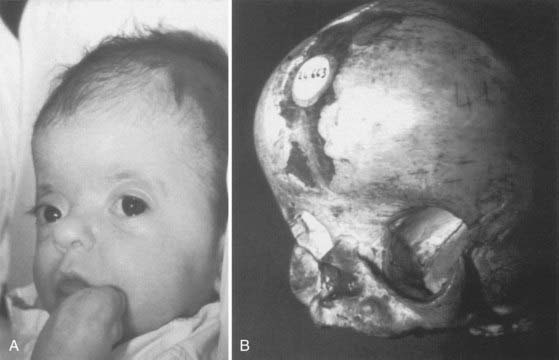
Described by Dr. Louis Edouard Octave Crouzon in 1912, this syndrome involves only the face and cranium and is not associated with other anomalies elsewhere on the limbs or trunk.1 The fundamental factor is an underdeveloped midfacial mass that features exorbitism because of the lack of depth of the orbits, inverted occlusion, and receding malar bones. The nose is short (Figs. 183-1 and 183-2). Cranially, brachycephaly is usually present, but sometimes scaphocephaly or a cloverleaf skull may be observed (Fig. 183-3).
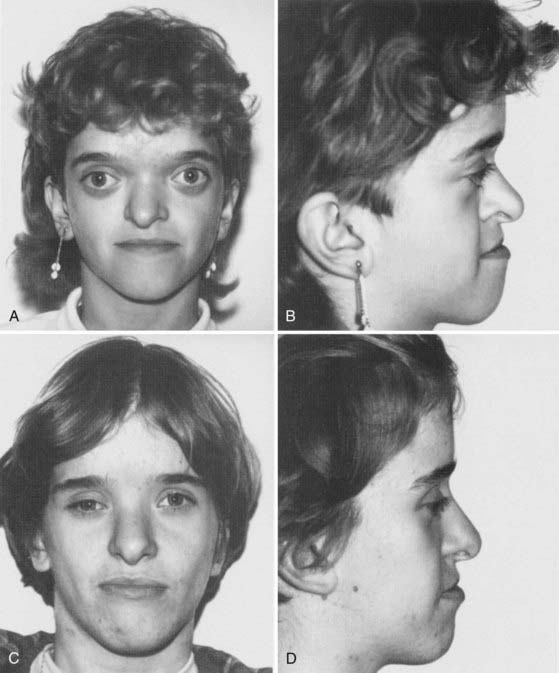
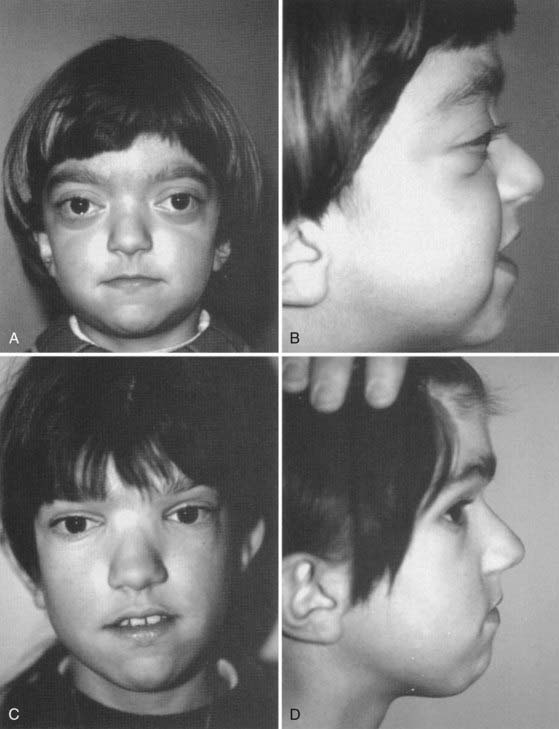
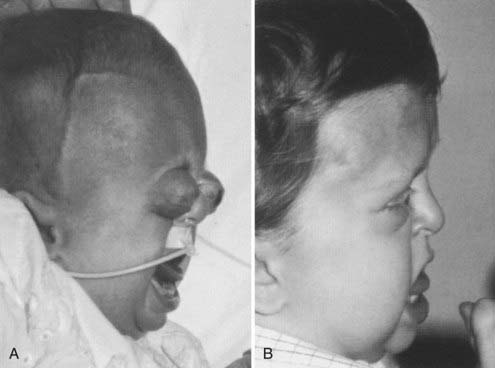
In some cases the diagnosis is evident at birth. In such infants the malformation is usually severe, with marked frontofacial retrusion producing severe exorbitism with a high risk for exposure keratitis or even subluxation of the globes. Retrusion of the maxillae is also severe and produces airway obstruction with obligatory mouth breathing (Fig. 183-4).
Apert’s Syndrome (Acrocephalosyndactyly)
First described by Apert in 1906, this syndrome is easy to recognize because of the associated syndactylies (fused digits) of the hands and feet.2 These syndactylies are always severe and affect almost all the digits. They can range from nearly complete fusion to fingers well delineated but joined by skin (Fig. 183-5).3
Pfeiffer’s Syndrome
Described by Pfeiffer in 1964, this syndrome is an association of faciocraniosynostosis and anomalies of the hands and feet.4–6 Although heterogeneous in manifestation, brachycephaly is present as a result of bicoronal synostosis (sometime asymmetric), midface retrusion secondary to maxillary hypoplasia, and hypertelorism. The thumbs and great toes are broad with a varus deviation. Soft tissue syndactyly can be observed. As in Crouzon’s and Apert’s syndromes, some severe forms exist, with precocious marked frontofacial retrusion resulting in ocular and breathing problems. This condition is sometimes associated with a cloverleaf skull.
Saethre-Chotzen Syndrome
Described by Saethre in 19317 and Chotzen in 1932,8 this syndrome is characterized by the association of bicoronal synostosis, maxillary hypoplasia, ptosis, and ear anomalies.5 The craniosynostosis is usually a brachycephaly or, in some cases, a plagiocephaly or even an oxycephaly (acrocephaly). The midface retrusion is most often mild. The ears have prominent antihelical crura. Some patients have soft tissue syndactyly in the hands.
Craniofrontonasal Dysplasia
In the group of patients with craniofacial dysplasia, some have a bicoronal craniosynostosis and form a subgroup with a condition called craniofrontonasal dysplasia, first described by Cohen in 1979.9 Brachycephaly, often marked, is associated with the facial anomalies of frontonasal dysplasia, which include hypertelorism, a broad nasal bridge, a bifid nose, and sometimes, soft tissue syndactyly.
Carpenter’s Syndrome
First described by Carpenter in 1901, this syndrome is characterized by craniosynostosis of varying sutures and variable polysyndactyly of the feet, brachydactyly of the fingers, and various soft tissue syndactyly.5 Additional abnormalities include congenital cardiac defects, short stature, obesity, and neurocognitive deficiency. Inheritance is autosomal recessive.
Etiology
Although the majority of faciocraniosynostoses are sporadic, there is also evidence for a genetic origin based on observations of familial cases and a growing body of molecular data10–16 (see also Chapter 181). Crouzon’s syndrome occurs in 1 in 25,000 births, and the frequency of familial cases varies in the literature from 44% to 67%.5 Familial cases accounted for 26% in the senior authors’ series. In this series (Table 183-1), the rate of familial cases was lower in the precocious forms of Crouzon’s syndrome than in the common type: 13% versus 29%, respectively. Transmission is autosomal dominant. There is great variability of expression, and both severe and mild forms can be observed in the same family. In fresh mutations (sporadic cases), the paternal age at conception is higher than the mean in the unaffected population. A continually increasing number of mutations for Crouzon’s syndrome have been identified, with most located on the third immunoglobulin-like loop of fibroblast growth factor receptor 2 (FGFR2).5
TABLE 183-1 Number of Patients Treated for Isolated Craniosynostosis and Craniofacial Syndromes in Our Series (1976-2004)
| TYPE | NO. OF PATIENTS TREATED |
|---|---|
| Nonsyndromic Craniosynostosis | |
| Oxycephaly | 115 |
| Trigonocephaly | 350 |
| Scaphocephaly | 799 |
| Lambdoids | 9 |
| Rare form | 93 |
| Positional plagiocephaly | 39 |
| Brachycephaly | 138 |
| Plagiocephaly | 335 |
| Total | 1878 |
| Syndromic Craniosynostosis | |
| Apert’s | 103 |
| Crouzon’s | 120 |
| Saethre-Chotzen | 79 |
| Pfeiffer’s | 43 |
| Frontonasal dysplasia | 30 |
| Other syndromes | 38 |
| Total | 413 |
The incidence of Apert’s syndrome is estimated to be between 1 in 100,000 and 1 in 160,000 births.5 Most cases are sporadic; few affected patients have children because of the severity of the disease. However, dominant transmission with complete penetrance has been reported in some cases. As in Crouzon’s syndrome, the paternal age at conception is higher than average. Most mutations associated with Apert’s syndrome are present in the linker region between IgII and IgIII on FGFR2.17
Pfeiffer’s syndrome has autosomal dominant transmission with complete penetrance and variable expression. In our series, 41% of the cases were familial. Mutations causing Pfeiffer’s syndrome are commonly found on FGFR1 and FGFR2.18,19
Saethre-Chotzen syndrome also has autosomal dominant transmission. Its penetrance is incomplete with variable expression. In our series, five of the nine cases were familial. In one of the first examples of molecular diagnosis of a craniofacial syndrome, the gene for Saethre-Chotzen syndrome was localized to the short arm of chromosome 7 (7p22) by Brueton and colleagues in 1992,20 and later the TWIST gene was implicated.21,22
Functional Aspects
Intracranial Pressure
More so than with isolated craniosynostoses, a problem of the faciocraniosynostoses is the associated risk for intracranial hypertension and its possible cognitive or visual sequelae. The risk for intracranial hypertension varies according to the type of syndrome.23–28 In the senior authors’ series, intracranial pressure was recorded in 68 patients with faciocraniosynostosis. Intracranial hypertension was defined as a baseline pressure of 15 mm Hg or greater. The frequency of intracranial hypertension was 62.5% in Crouzon’s syndrome, 45% in Apert’s syndrome, and 29% in the others.
In severe cases of multisuture synostosis, such as the Kleeblattschädel (cloverleaf) deformity, calvarial vault expansion may be required early because of problems with cranial constraint and elevated intracranial pressure. In such cases, posterior vault expansion can be performed as an initial first stage in neonates, with a second-stage fronto-orbital reconstruction and advancement being performed at the age of 4 to 9 months (see Fig. 183-3).
Mental Development
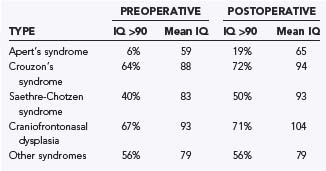
There is a great variability in neurocognitive functioning in the different types of faciocraniosynostosis.26,28,29 In comparing scores before and after surgery, three main factors appear to be involved as far as mental development is concerned. First, children with Apert’s syndrome have poorer cognitive development, and this is equally true before and after surgical treatment (Table 183-2). Second, early treatment leads to better cognitive outcomes (Table 183-3). Third, cognitive development is only mildly improved after treatment when compared with the patient’s preoperative status. In other words, the main predictive factor is preoperative cognitive status, which is best preserved by early frontal release.






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


