Chapter 67 The Cerebellum and the Hereditary Ataxias
Basic Cerebellar Structure, Function, and Dysfunction
Cerebellar Structure
The cerebellum can be divided anatomically in the anteroposterior and mediolateral dimensions (Figure 67-1A and B). Fissures divide the rostrocaudal length of the cerebellum into ten lobules, which are analogous to the gyri of the cerebral cortex. These lobules can be grouped into three lobes: the anterior lobe (lobules I–V), the posterior lobe (lobules VI–IX), and the flocculonodular lobe (lobule X) (Figure 67–1C). The mediolateral divisions are the vermis, which lies medially, and the hemispheres, which lie laterally and are separated from the vermis by shallow grooves.
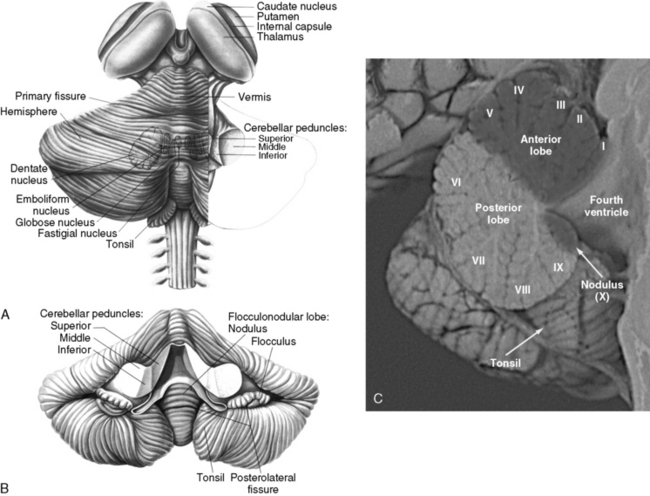
Fig. 67-1 The human cerebellum.
(A and B, From Kandel ER and Schwartz, JH, eds. Principles of neural science, 2nd edn. New York: Elsevier, 1985, pp 504 and 505. C, Courtesy of Dr. Dawna Armstrong.)
The cerebellar cortex is formed by five cell types, arranged in the same relative orientation, regardless of anatomical or functional location (Figure 67-2A). Purkinje cells are among the largest neurons in the central nervous system (CNS), and represent the sole output of the cortex. Cerebellar granule cells, which reside in the internal granular layer (IGL), are the most numerous neuronal population in the CNS. (Their numbers are nearly equal to all other CNS neurons combined.) Granule cells extend long, bifurcated axons (parallel fibers) into the molecular layer, where they travel for several millimeters and synapse with 1000–2000 Purkinje cells on their dendritic arbors; in turn, each Purkinje cell forms synapses with as many as 1,000,000 parallel fibers. Granule cells are the only excitatory cell type in the cerebellar cortex. Three local inhibitory interneurons also reside in the cortex. Golgi type II cells are large neurons found in the IGL that receive mossy fiber inputs and inhibit granule cells. Basket and stellate cells are found in the molecular layer, where they receive excitatory inputs from parallel fibers and are inhibitory to Purkinje cells.
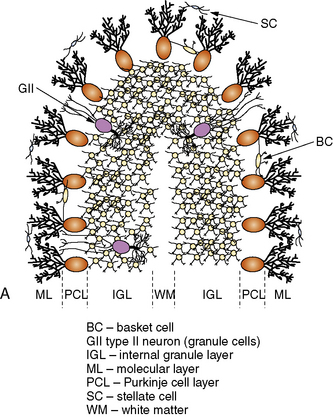
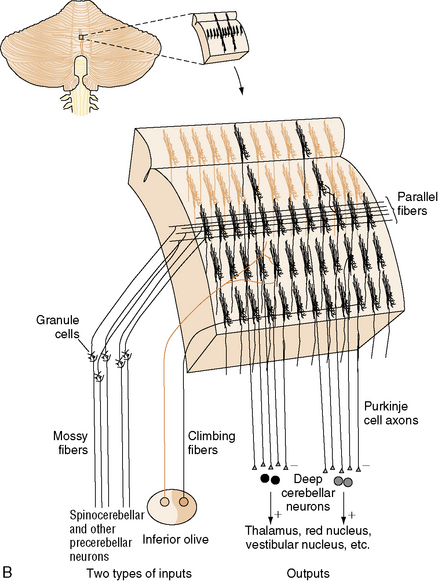
Fig. 67-2 Major cell types of the cerebellar cortex.
(B, From Kandel ER et al., eds. Principles of neural science, 4th edn. New York: McGraw-Hill, 2003, p 838.)
The neurons of the deep cerebellar nuclei (DCN) form four separate clusters in the cerebellar white matter (see Figure 67-1A). These cells receive inhibitory cortical input from the Purkinje cells, and excitatory extracerebellar afferents from climbing and mossy fibers. The fastigial nuclei lie most medially, and receive input from vermal Purkinje cells. The interposed nuclei (nucleus globose and nucleus emboliform) receive input from paravermal Purkinje cells. The dentate nuclei, which are the largest DCN in primates, receive input from the cerebellar hemispheres. Along with some Purkinje cells found in the flocculonodular lobe, the DCN represent the sole efferent output of the cerebellum, projecting to multiple brainstem nuclei in the medulla and pons, as well as the red nucleus and thalamus.
Cerebellar input consists of three classes of afferents. Climbing fibers originate in the inferior olives, which project to the contralateral cerebellar hemisphere. They provide strong excitatory input to Purkinje cells and neurons of the DCN. Each climbing fiber synapses on no more than ten Purkinje cells, while each Purkinje cell receives input from only one climbing fiber. Mossy fibers originate in multiple locations in the thalamus, brainstem, and spinal cord, and form excitatory synapses on granule cells and Golgi type II neurons in the IGL, as well as on neurons of the DCN. Climbing fibers, and to a lesser extent mossy fibers, are arranged in a series of parasagittal bands that “stack” next to one another in the mediolateral dimension (Figure 67-2B); the functional significance of this arrangement will be discussed in the next section. Finally, multilayer fibers originate in the hypothalamus, locus ceruleus, and raphe nuclei, and form a diffuse fiber network in the cortex and DCN.
Like the cerebral cortex, cerebellar cortical afferents and efferents are arranged in a somatotopic manner (reviewed in Manni and Petrosini [2004]). This organization is more complex than that of the cerebral cortex in two ways. First, at least three separate homunculi (one in the anterior lobe and one in each of the paravermian lobules) can be identified (Figure 67-3A). These are believed to converge on separate, continuous somatotopic maps in the four cerebellar nuclei. This arrangement, which was initially described in other species, has recently been directly demonstrated in humans using functional neuroimaging [Bushara et al., 2001; Manni and Petrosini, 2004]. Second, the sensory map within a cerebellar folium is not continuous. Instead, a fractured somatotopy exists, where each region of the face or limb has multiple representations within the folium that may not be adjacent to one another (Figure 67-3B). The reason for this arrangement is unknown, but it may be necessary to bring climbing fiber and mossy fiber inputs into register with one another.
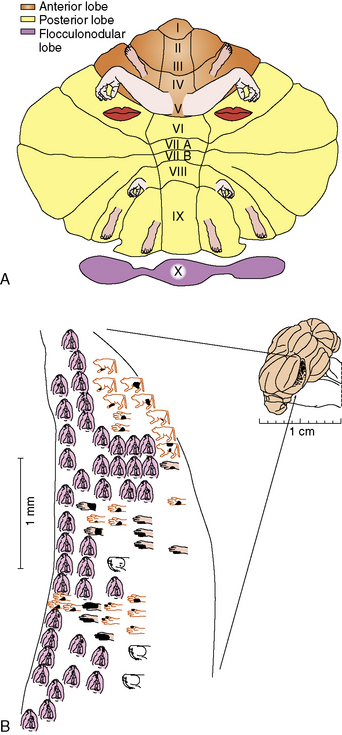
Fig. 67-3 Somatotopic organisation of the cerebellum.
(A, From Manni E and Petrosini L. A century of cerebellar somatotopy: A debated representation. Nature Reviews of Neuroscience 2004;5:241–249. B, King JS, ed. Neurology and neurobiology, vol. 22. New York: AR Liss, 1987.)
Cerebellar Function
As noted above, cerebellar afferent input is segregated into a series of parasagittal bands. There are two main hypotheses as to how this parasagittal arrangement might be functionally important. First, comparison of climbing fiber to mossy fiber excitatory inputs within these bands could contribute to refining movements and motor learning [Ito, 2002]. It has also been proposed that mossy fiber input, which is translated through granule cells and their parallel fibers, could transform spatial relationships into temporal ones as the signal passes from Purkinje cells closest to the granule cells to those farther down the parallel fiber path. The receptive Purkinje cells could then detect timing differences between the incoming granule cell and climbing fiber signals. Thus, the cerebellum could act as an alarm clock, stopwatch, or coincidence/time shift detector [Braitenberg, 1967].
Positron emission tomography (PET) and functional magnetic resonance imaging (fMRI) suggest that the cerebellum is involved in several nonmotor functions. These include sensory discrimination, attention, working memory, semantic association, verbal learning and memory, and complex problem solving [Allen et al., 1997]. A number of other studies have also linked the cerebellum to various higher-order cognitive processes [Leiner et al., 1991; Middleton and Strick, 1994]. Most of these functions are subserved by the cerebellar hemispheres and the dentate nuclei.
Cerebellar Dysfunction
Cerebellar (ataxic) dysarthria produces abnormalities in articulation (slurring and inaccuracy in range, force, and timing; patients sound as if inebriated), phonation (vocal quality can be harsh or uneven), resonation (a nasal quality is common), and prosody (patients tend to place equal and excessive emphasis on each syllable). Other kinds of dysarthria exist; there is, for example, the hypokinetic dysarthria seen in Parkinson’s disease, in which bradykinesia due to loss of dopaminergic neurons in the substantia nigra tends to produce low volume of speech or hoarseness, reduced pitch variability, palilalia (repetition of syllables), and very slow speech. There is evidence that left cerebellar lesions interfere with speech prosody because of disruptions of interconnections with the right cerebral hemisphere, which mediates this process [Brazis et al., 2001]. Cerebellar mutism is sometimes seen after removal of vermian tumors, and may result from bilateral involvement of the dentatorubrothalamic tracts.
Nonmotor manifestations of cerebellar disease can also occur. A cerebellar cognitive affective syndrome has been described in patients with isolated cerebellar lesions of a number of different etiologies [Schmahmann and Sherman, 1998]. In this syndrome, executive functions, spatial cognition, personality changes, and language deficits are all present. The postulated neural substrate involves circuits that link prefrontal, posterior parietal, superior temporal, and limbic cortices to the cerebellum.
Differential Diagnosis of Ataxia
The underlying cause of ataxia is virtually always primary or secondary dysfunction of the cerebellum. Cerebellar disease is a result of a number of underlying conditions, many of which are listed in Box 67-1. The most prevalent causes of acute cerebellar ataxia are viruses (e.g., coxsackievirus, rubeola, varicella), toxins (e.g., alcohol, barbiturates, antiepileptic drugs), and traumatic insults. Tumors of the cerebellum (vermal and hemispheric) or the adjacent brainstem, particularly pontine gliomas, are also relatively common. Numerous metabolic conditions affecting the central and peripheral nervous systems lead to ataxia. Congenital abnormalities of the nervous system, such as Chiari malformation, Dandy–Walker malformation, and basilar impression, are associated with ataxia. Endocrinologic abnormalities, particularly hypothyroidism, may also present with ataxia as the predominant manifestation. Hemorrhage, infarction, and embolism are unusual causes of cerebellar damage and often occur in conjunction with damage to neighboring brain regions. Angioblastomas of the cerebellum are rare; von Hippel–Lindau disease, a neurocutaneous condition, is associated with vascular lesions of the cerebellum, as is the PHACE (posterior fossa malformations, hemangiomas, arterial anomalies, coarctation of aorta and cardiac defects, and eye abnormalities) syndrome [Frieden et al., 1996].
Box 67-1 Selected Causes of Ataxia in Childhood










Degenerative and/or Genetic

 Ataxia, retinitis pigmentosa, deafness, vestibular abnormality, and intellectual deterioration [Francois and Descampas*]
Ataxia, retinitis pigmentosa, deafness, vestibular abnormality, and intellectual deterioration [Francois and Descampas*]

 Cerebellar ataxia with deafness, anosmia, absent caloric responses, nonreactive pupils, and hyporeflexia [Brown, 1959]
Cerebellar ataxia with deafness, anosmia, absent caloric responses, nonreactive pupils, and hyporeflexia [Brown, 1959]



 Hereditary cerebellar ataxia, intellectual retardation, choreoathetosis, and eunuchoidism [Altchule and Kotowski*]
Hereditary cerebellar ataxia, intellectual retardation, choreoathetosis, and eunuchoidism [Altchule and Kotowski*]




 Periodic attacks of vertigo, diplopia, and ataxia – autosomal-dominant inheritance [Farmer and Mustian*]
Periodic attacks of vertigo, diplopia, and ataxia – autosomal-dominant inheritance [Farmer and Mustian*]













































































Management of Cerebellar Dysfunction and Ataxia
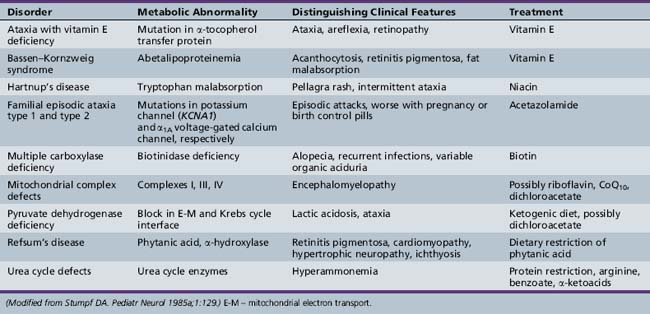
When ataxia is the result of certain metabolic disorders or toxins, trauma, or neoplasia, specific therapies for the underlying condition can be curative (Table 67-1). Direct neurological causes of ataxia, however, such as inherited diseases, are not yet amenable to treatment. Patients can benefit from various aids for ambulation, as well as physical therapy and occupational therapy; though these approaches will not restore mobility in advanced disease, they none the less alleviate other symptoms such as spasticity and can greatly improve quality of life.
The Hereditary Ataxias
The classification of the hereditary ataxias has long posed a clinical challenge. Prior to the advent of gene cloning technology, these conditions were classified based on their mode of inheritance, clinical features, and pathological findings. These approaches ultimately proved to be unsatisfactory, and often led to more confusion than clarity. Patients with known hereditary ataxia have a range of cerebellar dysfunction that may be exclusively cerebellar in origin or combined with other features, including brainstem dysfunction, spinal cord abnormalities, extrapyramidal findings, neuropathy, retinopathy, deafness, cataracts, seizures, and/or dementia [Greenfield, 1954; Koeppen and Barron, 1984; Zoghbi et al., 1993]. The age of onset and clinical manifestations of any given ataxia may differ significantly from family to family and even in the same kindred, making specific assignment beyond mode of inheritance difficult [Currier et al., 1972]. Likewise, attempts to classify ataxia according to the anatomic site of the most dominant pathology (e.g., spinal cord, cerebellum) are also flawed. Descriptors such as “olivopontocerebellar atrophy” (OPCA), a pathological finding seen most predominantly in the dominantly inherited ataxias, do not adequately serve to distinguish between them.
Autosomal-Recessive Inherited Syndromes
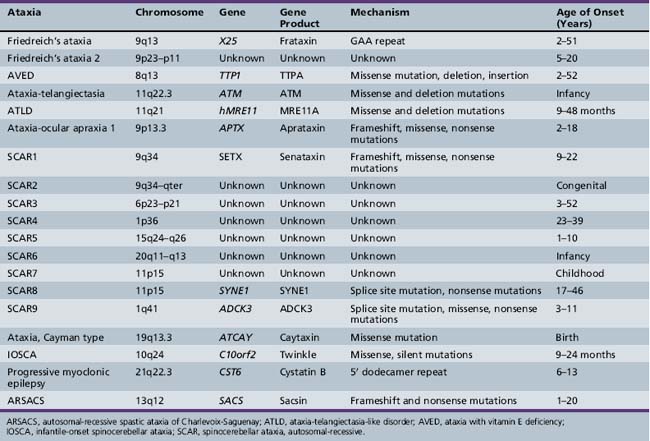
The autosomal recessive inherited ataxias (summarized in Table 67-2) are a varied group of disorders that tend to have an early age of onset in infancy or childhood. They can involve multiple regions of the central and peripheral nervous system. In general, earlier age of onset heralds a more aggressive disease course. Several causative mutations have been identified, leading to a clear genetic classification scheme.
Friedreich’s Ataxia (Freidreich’s Ataxia; Spinocerebellar Ataxia – OMIM 229300)
Friedreich ataxia is one of the most common hereditary ataxias, with a prevalence of approximately 1–2 per 100,000 in the general population [Harding, 1984]. Caucasians are affected more often than other racial groups, with a prevalence closer to 1 per 50,000 [Cossee et al., 1997]. Clinically, the disease is signaled by the presence of ataxia and corticospinal tract dysfunction. Other associated features include nystagmus, kyphoscoliosis, and an unusual cardiomyopathy. The disease was originally reported by Friedreich [Friedreich, 1863], who believed it was associated only with spinal cord abnormalities, subsequent findings demonstrated involvement of the medulla, peripheral nerves, and cerebral cortex in some cases.
Clinical manifestations
Loss of cells in cranial nerves VII, X, and XII causes facial weakness, with cerebellar dysarthria and dysphagia. Lateral or horizontal nystagmus may be prominent, and visual pursuit is punctuated with small jerks. Impairment of pupillary responsivity and limited extraocular movements are also common. Additional ophthalmologic abnormalities include cataracts, retinitis pigmentosa, and optic atrophy. Auditory dysfunction is common [Cassandro et al., 1986], and is documented by abnormal brainstem auditory-evoked potentials that are likely the result of impairment of both central and peripheral acoustic pathways [Knezevic and Stewart-Wynne, 1985]. The degree of hearing compromise is variable; in addition, some patients experience vertigo. Although higher cortical function appears intact by clinical measures, some studies suggest a decrease in information processing speed [Hart et al., 1985].
The lower limbs typically show loss of deep tendon reflexes, but reflexes may be preserved for a longer period in young children [Salih et al., 1990], and patients with late-onset disease can be spastic with hyperreflexia. Bilateral extensor toe signs are easily elicited. Cremasteric and abdominal reflexes may be lost. The masseter reflex remains intact [Auger, 1992]. Occasionally, weakness, muscle atrophy, and profound hypotonia may be evident, usually associated with spinal nerve disintegration. Virtually all facets of cerebellar function are involved, and both gross and fine coordination are affected. Position sense, vibration sense, and other skills requiring normal posterior column function are grossly impaired. Autonomic innervation of superficial layers of skin may be involved. Pain, touch, and temperature sensation are usually intact, but diminish over the course of the illness. Evidence of axonal sensory neuropathy, indicated by electrodiagnostic studies, is invariably present. Somatosensory-evoked potentials are abnormal, as is central motor function, as determined by magnetic stimulation [Claus et al., 1988].
Patients with Friedreich’s ataxia often develop talipes equinovarus or pes cavus. Deformities of the hand can also occur. Flexor spasms can become a great source of pain. Many patients suffer from kyphoscoliosis [Labelle et al., 1986], and may be unable to walk secondary to poor balance and an abnormally shifted center of gravity (Figure 67-4). Surgical intervention can correct the abnormal spinal curvature, and this is likely to help with ventilation and autonomic functions, even if it does not restore ambulation.

Fig. 67-4 Friedreich’s ataxia and moderate scoliosis in a 13-year-old boy who is unable to walk independently.
Cardiomyopathies are the most common accompanying cardiac abnormalities, although congenital heart disease has been described on occasion. The cardiomyopathy is often progressive, and is documented even before clinical manifestations by the presence of electrocardiogram (EKG) abnormalities (e.g., deep Q waves, low-wave QRS complexes, S-T segment changes, T-wave inversion) [Harding and Hewer, 1983; Child et al., 1986]. Arrhythmias may be particularly worrisome and life-threatening. EKG and vector cardiographic changes are demonstrable in more than 90 percent of patients [Child et al., 1986]. Left ventricular hypertrophy or symmetric ventricular hypertrophy is frequently evident on echocardiography [Pentland and Fox, 1983]. Hypertrophic cardiomyopathy is the leading cause of death in Friedreich’s ataxia patients.
Diabetes mellitus is associated with Friedreich’s ataxia; even in its absence, the glucose tolerance curve may be abnormal. Insulin resistance appears to be the explanation [Khan et al., 1986]. Unaffected relatives may also manifest insulin resistance. Episodic hypothermia, intermittent vomiting, and ventilatory dysfunction have also been reported [Thoren, 1962].
Imaging studies show both structural and functional changes that correlate with pathologic progression. MRI studies document atrophy of the upper cervical spinal cord and cerebellum [Wessel et al., 1989]. PET imaging demonstrates a widespread increase in cerebral metabolic rate for glucose in the brain of ambulatory patients with Friedreich’s ataxia. As the disease progresses, glucose metabolism decreases in a regionally specific manner; only the caudate and lenticular nuclei have increased metabolic rate in nonambulatory patients [Gilman et al., 1990].
Diagnosis is suggested by the presence of the expected clinical manifestations and sometimes by family history. Distinct diagnostic criteria for Friedreich’s ataxia were advanced by Geoffroy and colleagues [Geoffroy et al., 1976]: autosomal-recessive inheritance, onset before 10 years of age, gait ataxia, dysarthria, absent deep tendon reflexes, dorsal column signs, and weakness. The presence of an abnormal 5-hour glucose tolerance curve is corroborative. Spinal fluid findings may include elevated protein content and mild pleocytosis.
Pathology
The primary site of involvement on pathologic examination is the spinal cord. Gross examination reveals a shrunken cord, with the posterior columns, spinocerebellar tracts, corticospinal tracts, and posterior rootlets strikingly affected by varying degrees of fiber loss, demyelination, and gliosis. Conditional Freidreich’s ataxia gene inactivation in experimental animals showed that anomalies observed in the DRG (dorsal root ganglion) neurons are primary events, whereas neuronal loss in the posterior thoracic nucleus (Clarke’s column) and degeneration in the posterior columns may be secondary events [Simon et al., 2004]. Axons and myelin alike are involved, although the medullary nuclei (Burdach’s nuclei) are usually spared [Greenfield, 1954]. Structures in the medial root zone, including ganglion cells and the posterior roots that terminate within them, manifest varying degrees of involvement but are typically decreased in number. The lumbar and sacral roots are most severely affected, and the large, myelinated lumbar spinal nerves lose many axons. In contrast, anterior horn cells and their rootlets are usually uninvolved; however, minor pathologic alterations may be evident. Myopathic changes are rare.
The density of large myelinated fibers in sural nerve biopsies is reduced, even in young children, and often worsens with age [Said et al., 1986].
Pathologic changes in the heart include evidence of cardiomyopathy and involvement of blood vessels, nerves, and ganglia [James et al., 1987].
Genetics
Friedreich’s ataxia is transmitted as an autosomal-recessive disorder; the finding of consanguinity among several families clearly supported the autosomal-recessive inheritance pattern. The disease affects males and females equally. The establishment of the criteria proposed by Geoffroy and co-workers [Geoffroy et al., 1976] provided a basis for research studies and allowed clinical identification of families for linkage studies. Genetic linkage studies mapped the Friedreich’s ataxia gene to chromosome 9 and confirmed that the “Acadian” form of ataxia, characterized by a slower course of degeneration [Barbeau et al., 1984; Keats et al., 1989], also maps to chromosome 9 [Chamberlain et al., 1988; Keats et al., 1989]. Friedreich’s ataxia is caused by decreased expression of the mitochondrial protein, frataxin, encoded by the X25 gene located at 9q13 [Bidichandani et al., 1998; Koeppen, 1998].
Campuzano et al. [1996] were the first to identify an unstable GAA trinucleotide repeat in the first intron of X25. They also identified three different point mutations. In the group of 74 patients without a point mutation, 71 were homozygous for expanded alleles, and 3 were heterozygous for the expanded repeat. Five patients with point mutations were heterozygous for the expansion. The size of the repeat ranged from 7 to 22 on normal alleles and from 200 to 900 on Friedreich’s ataxia chromosomes. Further studies revealed that the triplet repeat could get as large as 1700 units, and that the lengths of both the larger and the smaller expanded alleles correlated inversely with the age of onset and severity of disease [Durr et al., 1996; Filla et al., 1996]. The mean allele length is significantly higher in Friedreich’s ataxia patients with diabetes, cardiomyopathy, and loss of reflexes in the upper extremities. Of 187 patients with autosomal-recessive ataxia, 140 were homozygous for a GAA expansion [Durr et al., 1996]. Furthermore, age-dependent accumulation of large GAA expansions contributes to the progressive pathology involving in the DRGs. Therefore, somatic instability of the expanded GAA triple repeat sequence may contribute directly to disease pathogenesis and progression in specific tissues [De Biase et al., 2007].
Frataxin is localized to the inner mitochondrial membrane and is thought to play a role in mitochondrial respiration and iron accumulation. Deletion of the frataxin homolog in yeast leads to mitochondrial iron accumulation and increased susceptibility to oxidative stress, with an increased production of free radicals (reviewed in Voncken et al. [2004]). These observations, coupled with the fact that expanded trinucleotide repeats interfere with transcription elongation of the X25 gene and cause a frataxin deficiency state, might explain the human phenotype [Bidichandani et al., 1998]. The high evolutionary conservation of frataxin has enabled the development of disease models in various organisms, from the unicellular eukaryote Saccharomyces cerevisiae to the mouse. Yeast was an important model system for identifying the crucial role of frataxin in the regulation of intracellular iron trafficking, iron-sulfur (Fe-S) cluster and heme biogenesis, and oxidative metabolism. Several Caenorhabditis elegans models with knockdown of the C. elegans FXN homolog frh-1 show conflicting results in response to oxidative stress, but all of them show reduced life span and may be useful for drug screening in Freidreich’s ataxia. Moderate reduction of the Drosophila frataxin homolog (dfh) causes a hypersensitive response to hypoxia, which upholds a causative role for oxidative stress in Freidreich’s ataxia. Mouse models of Freidreich’s ataxia have provided the most insight, however (reviewed in Puccio [2009]). The knockout model of frataxin suffers early embryonic lethality without iron accumulation, while heterozygosity for the frataxin null allele shows no pathological phenotype. These results suggest that residual frataxin expression associated with expansion mutation is critical for survival [Cossee et al., 2000]. Conditional animal models are viable and reproduce certain morphological and biochemical features observed in patients with Freidreich’s ataxia, including cardiac hypertrophy without skeletal muscle involvement, large sensory neuron dysfunction without alteration of small sensory and motor neurons, and deficient activities of complexes I through III of the respiratory chain and of the aconitases, enzymes essential in the citric acid cycle (reviewed in Puccio [2009]). Humanized GAA repeat expansion mouse models were obtained by combining the constitutive knockout model with the transgenic expression of a yeast artificial chromosome (YAC) carrying the human locus. These animals exhibit oxidative stress, leading to progressive neuronal and cardiac pathology [Al-Mahdawi et al., 2006]. Although none of these mouse models is authentic, a multimodel approach facilitates the study of different pathophysiological mechanisms and the examination of different therapeutic strategies. Why only selective neuronal systems are vulnerable to the loss of frataxin function remains unknown.
Treatment
Given the proposed function of frataxin protein in iron-sulfur cluster biosynthesis and the increased oxidative stress found with decreased protein levels, several groups have initiated treatment with idebenone, a short-chain analog of coenzyme Q10 (reviewed in Schulz et al. [2009]). All but one of these trials (the shortest trial) with idebenone at 5 mg/kg and 10 mg/kg/day have demonstrated improved cardiac function and decreased ventricular mass: important findings, since cardiac dysfunction is the leading cause of death in Friedreich’s ataxia patients. In these low-dose trials, idebenone treatment has no effect on the progression of ataxia [Schulz et al., 2009], but Di Prospero et al. showed that higher doses of idebenone (up to 60 mg/kg/day) are well tolerated, provide neurological benefit, and improve activities of daily life (ADL) in Freidreich’s ataxia patients who are still ambulatory [Di Prospero et al., 2007].
A high proportion of Freidreich’s ataxia patients have decreased serum CoQ10 levels, which was the best predictor of a positive clinical response to CoQ10/vitamin E therapy. Low- and high-dose CoQ10/vitamin E therapies were equally effective in improving ICARS scores (International Cooperative Ataxia Rating Scale) [Cooper et al., 2008]. Therapy with other antioxidants, iron chelators, and/or glutathione peroxidase mimetics has been suggested (and in some cases tried), but no clear beneficial effect has been established.
Expansion of intronic GAA repeat likely induces chromatin changes and in turn FXN silencing in Freidreich’s ataxia [Herman et al., 2006]. Treatment with a histone deacetylase (HDAC) inhibitor restored wild-type frataxin levels in the nervous system and heart, and increased histone H3 and H4 acetylation in chromatin near the GAA repeat [Rai et al., 2008]. These findings provide a very promising prospect for Freidreich’s ataxia treatment using HDAC inhibitors.
Friedreich’s Ataxia 2 (OMIM 601992)
Other families with a Friedreich’s ataxia disease phenotype, but without point mutations or GAA expansions in the X25 gene, have been described [Kostrzewa et al., 1997; Christodoulou et al., 2001]. In one family, linkage to a locus on chromosome 9p23–p11 has been found [Christodoulou et al., 2001]. The biological basis for the genocopy is not known, but it has been proposed that mutations in genes that lie in the iron metabolism pathway may be responsible.
Vitamin E Deficiency and Related Syndromes
Numerous mutations have been identified in the tocopherol transfer protein gene (TTP1), which encodes the α-tocopherol transfer protein (TTPA) [Ouahchi et al., 1995; Cavalier et al., 1998]. TTPA is expressed in the liver and is responsible for the incorporation of tocopherol (vitamin E) into very-low-density lipoprotein (VLDL) [Traber et al., 1990]. Mutated TTPA proteins fail to facilitate the secretion of vitamin E from cells, and the severity of the AVED pathology corresponds to the degree of impairment associated with each mutation [Qian et al., 2006].
Ataxia-Telangiectasia (AT; Louis-Bar Syndrome – OMIM 208900)*
The initial clinical description of this disease was reported by Syllaba and Henner [1926], who called it congenital double athetosis-aconjunctival vascular plexus syndrome. Louis-Bar [1941] reported a 9-year-old male with associated ataxia and oculocutaneous telangiectasias, classifying the case as a previously unrecognized phakomatosis. Two additional independent reports subsequently appeared in the literature [Biemond, 1957; Boder and Sedgwick, 1957], and Boder and Sedgewick named the condition ataxia-telangiectasia.
Clinical manifestations
Early motor development appears to be normal until around the time that the child starts walking, when ataxia is noted. The ataxia is progressive and ultimately leads to an inability to ambulate by the beginning of the second decade. Choreoathetosis and dystonia occur in up to 90 percent of patients, and these motor findings become more prominent with increasing age. Facial weakness leads to the characteristic impassive faces, as well as drooling and dysarthria. While strength is initially normal, many patients in their 20s and 30s develop progressive spinal muscular atrophy, predominantly affecting the hands and feet [Gatti et al., 1991]. Peripheral neuropathy in the form of diminished deep tendon reflexes and loss of large fiber sensation is also seen. Mental function is well preserved, although deficits in short-term memory can occur in the third and fourth decades [Gatti et al., 1991].
Oculomotor apraxia is a distinguishing feature of the disease. The apraxia commonly presents before the appearance of conjunctival telangiectasias and is characterized by defects of initiating voluntary saccades, hypometric voluntary saccades accompanied by compensatory eye-blinking and/or head-thrusting movements, and disrupted smooth pursuit movements [Smith and Cogan, 1959; Baloh et al., 1978]. Involuntary saccade initiation and optokinetic nystagmus may be impaired as well, and oculovestibular reflexes may be increased.
Telangiectasias are usually first observed in patients between the ages of 2 and 4 years, although they can occur as early as birth and as late as 14 years of age [Centerwall and Miller, 1958; McFarlin et al., 1972] (Figure 67-5). In addition to the conjunctivae, they appear on exposed areas of the skin, particularly areas of friction and trauma, such as the auricle, nasal bridge, and antecubital and popliteal spaces. Exposure to sun enhances their appearance. Premature aging of hair and skin is frequent, as are skin infections, including chronic blepharitis. Other skin changes, consisting of vitiligo and café-au-lait spots, can be seen. Rarely, scleroderma-like lesions occur.
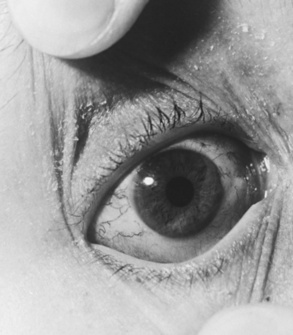
Fig. 67-5 Characteristic conjunctival telangiectasias observed in a 12-year-old with ataxia-telangiectasia.
Recurrent sinopulmonary infections are common, affecting 90 percent of patients, and usually result in chronic bronchitis, bronchiectasis, or both. The impairment of cellular immunity is also manifest by abnormally developed or absent adenoids, tonsils, lymphoid tissue, and thymus gland. Patients have an impaired delayed hypersensitivity response to skin-sensitizing antigens and a delayed homograft-rejection response [McFarlin and Oppenheim, 1969].
Patients with AT are prone to develop tumors and are more likely than the general population to have Hodgkin’s disease, leukemia, lymphoma, and lymphosarcoma. Other associated neoplasms include brain tumors, gastric adenocarcinomas, ovarian dysgerminomas, gonadoblastomas, cystic adenofibromas, uterine leiomyomas, and thyroid adenomas [Miller and Chatten, 1967; Gatti and Good, 1971].
Patients with less severe variant forms of AT have been described [Gilad et al., 1998; Saviozzi et al., 2002]. Immunodeficiency, telangiectasias, cancer, and sinopulmonary infections may be absent or reduced, but the neurological manifestations still occur. These individuals have a later onset and slower progression of neurological signs, longer life spans, and decreased chromosomal instability and cellular radiosensitivity.
Laboratory findings
Fibroblast and lymphoid cell lines from AT patients show increased radiosensitivity, leading to spontaneous and radiation-induced chromosomal breakage and rearrangement, which can be assessed in culture [Shiloh et al., 1985]. In lymphoid cell lines, the chromosomal breaks occur at the loci of T-cell antigen receptors and immunoglobulin genes, where DNA rearrangements and deletions naturally occur [Hecht and Hecht, 1985].
Pathology
Cerebellar atrophy primarily affects the Purkinje and granular cells, although basket cells can also be involved. Neuronal degeneration of the dentate and olivary nuclei and the substantia nigra has been demonstrated, and nuclear changes occur in the cells of the oculomotor complex, pretectal nuclei, and hypothalamus. Pituitary cells often have enlarged or dysplastic nuclei. Older patients have denervation of the posterior columns of the spinal cord. Degeneration of the anterior horn cells, decreased numbers of satellite cells in the dorsal root ganglia, and nucleomegaly of Schwann cells have been described [De Leon et al., 1976].
Genetics
The worldwide incidence of AT is approximately 1 in 40,000–100,000 live births; the carrier frequency ranges from 0.5 to 1.0 percent. The gene locus maps to chromosome 11q22–23 [Gatti et al., 1988]. The ATM (ataxia telengiectasia mutated) gene is very large, with 66 exons spanning 150 kb of genomic DNA [Uziel et al., 1996]; this fact makes mutation detection a challenge. In addition, because of the large number of different mutations, the majority of patients are compound heterozygotes. Still, using a combination of techniques, the estimated mutation detection rate has been detected to be over 95 percent in affected individuals [Buzin et al., 2003]. It is estimated that truncating mutations account for about 85 percent of mutations, while point mutations are responsible for the rest [Buzin et al., 2003].
ATM is a nuclear phosphoprotein homologous to a family of phosphatidylinositol kinase-related proteins, and it functions in the DNA damage response and in cell cycle regulation [Savitsky et al., 1995a, b]. Individuals with classic AT have little or no active ATM protein, while most individuals with variant AT have one severe mutation coupled with a mild mutation. In this latter condition, some ATM activity is preserved, which probably explains the milder phenotype [Saviozzi et al., 2002].
Loss of ATM activity could lead to neuronal degeneration in at least two ways. As ATM is a monitor of DNA strand breaks important for initiating repair, it has been suggested that ATM is necessary for the elimination of neural cells with genomic damage, and that the preservation of those cells may contribute to neuronal dysfunction [Herzog et al., 1998]. Several other diseases that affect DNA repair (ataxia-telangiectasia-like disorder, ataxia-ocular apraxia 1, xeroderma pigmentosum, Cockayne’s syndrome, Nijmegen breakage syndrome, and de Sanctis–Caccione syndrome) have a cerebellar phenotype, suggesting that cerebellar neurons may be very sensitive to DNA damage. This hypothesis highlights a nuclear role for the ATM protein. In neurons, however, a large percentage of the protein is found in the cytoplasm and axons. Increased numbers of lysosomes are found in Purkinje cell bodies and axons in ATM-deficient mice [Barlow et al., 2000]. This, coupled with an observed interaction with β-adaptin, a protein important for clathrin-mediated endocytosis, suggests that vesicle/protein transport may also be disrupted in ATM-defective cells [Lim et al., 1998].
Treatment
AT is a multisystem disease and its treatment requires attention to multiple types of interventions. Currently, we are not able to halt progressive neurodegeneration, although some features of the disease might respond to treatment. l-DOPA derivatives and anticholinergics may improve basal ganglia dysfunction. Amantadine, fluoxetine, or buspirone may help with the loss of balance and impaired speech; furthermore, tremors can often be controlled by gabapentin, clonazepam, or propranolol (reviewed in Lavin et al. [2007]).
Patients should receive vigorous supportive therapy, with particular attention directed to recurrent sinopulmonary infection. Attempts to improve immunologic status by plasma transfusion of thymosin and provision of fetal thymus transplants have not altered the course of the neurologic signs and symptoms [Wara and Ammann, 1978]. Treatment of neoplasms is a delicate proposition because patients are extremely sensitive to radiation and chemotherapy, and demonstrate resultant ulcerative dermatitis, severe esophagitis, dysphagia, and deep-tissue necrosis [Gotoff et al., 1967].
Ataxia-Telangiectasia-Like Disorder (ATLD – OMIM 604391)
ATLD patients have been described with clinical presentations including cerebellar ataxia and oculomotor apraxia, which are essentially identical to that of ataxia-telangiectasia except that telangiectasias are absent [Klein et al., 1996; Fernet et al., 2005]. Two of the patients, who were brothers, presented first with chorea that later progressed to ataxia [Klein et al., 1996]. In 2005, Fernet et al. described ten ATLD patients from three unrelated Saudi Arabian families. These patients had slowly progressive ataxia and ocular apraxia, but they did not develop tumors. Furthermore, they did not have telangiectasia, raised AFP, or reduced immunoglobulin levels [Fernet et al., 2005].
Increased susceptibility to DNA damage was noted in all four patients. Stewart et al. [1999] showed that the disorder maps to chromosome 11q21 in all four patients. Loss-of-function mutations in the hMRE11A gene are causative. Like ATM, MRE11A is important for DNA repair, and may act as a sensor of DNA damage. Stewart et al. estimate that approximately 6 percent of AT patients may in fact have ATLD [Stewart et al., 1999].
Ataxia-Oculomotor Apraxia 1 (AOA1; Early-Onset Ataxia with Ocular Motor Apraxia and Hypoalbuminemia – OMIM 208920)
This autosomal-recessive ataxia was initially described in Japanese families but has subsequently been described in other areas of the world as well [Tranchant et al., 2003]. It is the most frequent cause of autosomal-recessive cerebellar ataxia in Japan, and the second most common cause in Portugal. The presentation is nearly identical to that of AT without the non-neurological features [Aicardi et al., 1988], and accounts for up to 10 percent of autosomal-recessive cerebellar ataxias. Gait imbalance and dysarthria are the typical presenting features, although chorea can sometimes be the presenting symptom. Ocular apraxia typically occurs a few years after symptom onset; with time, progressive ophthalmoplegia occurs as well. Areflexia, dystonia, choreoathetosis, sensory and motor neuropathy, cognitive impairment, and retinal/macular lesions seen on funduscopy can also be part of the presentation. Life span is not affected.
Laboratory studies reveal hypoalbuminemia and hypercholesterolemia in the majority of cases as a later manifestation of the disease. Neuroimaging demonstrates cerebellar and sometimes brainstem atrophy. Pathology studies show loss of Purkinje cells in the cerebellum, and degeneration of the posterior columns, spinocerebellar tracts, and anterior horn cells of the spinal cord [Sekijima et al., 1998].
The gene, APTX, maps to chromosome 9p13.3 and is expressed in all body tissues [Date et al., 2001; Moreira et al., 2001]. Several mutations have been described, as have compound heterozygotes; frameshift and nonsense mutations result in more severe phenotypes than missense mutations. The APTX gene product, aprataxin, has domains that suggest a potential role in single-strand DNA repair, and has been shown to interact with another protein implicated in this process [Sano et al., 2004]. CoQ10 is often low in patients with AOA1 [Le Ber et al., 2007], and this deficiency responds to CoQ10 supplementation [Quinzii et al., 2005].
Spinocerebellar Ataxia, Autosomal-Recessive 1 (SCAR1 – OMIM 606002); Ataxia-Oculomotor Apraxia 2 (AOA2)
A second AT-like spinocerebellar ataxia with slightly different features than AOA1 has been described in families of several different ethnic backgrounds. The clinical features of AOA2 include gait ataxia, sensorimotor neuropathy, and ocular apraxia, the latter occurring about 50 percent of the time. While overall cognitive function is normal, subtle changes in executive function can be seen on neuropsychologic testing [Le Ber et al., 2004]. Neuroimaging demonstrates cerebellar atrophy. AOA2 is distinguished from AOA1 by the later age of onset, less common ocular apraxia, high levels of serum AFP, and normal serum albumin. The functional prognosis is also better in AOA2. The gene (SETX) maps to chromosome 9q34 [Nemeth et al., 2000]. Mutations cause premature termination of the senataxin protein, which is a member of the helicase family and is involved in RNA maturation and termination [Moreira et al., 2004]. Recently, Suraweera et al. [2009] have proposed a role for senataxin in the regulation/modulation of transcription.
Interestingly, other mutations in senataxin are transmitted in an autosomal-dominant manner and cause juvenile amyotrophic lateral sclerosis (ALS4) [Chen et al., 2004a]; the basis for the dramatic phenotypic difference between the two diseases is unknown.
Spinocerebellar Ataxia, Autosomal-Recessive 2 (SCAR2 – OMIM 213200); Cerebellar Hypoplasia, Nonprogressive Norman Type
Norman [1940] described three siblings in one family and two siblings in another who had had cerebellar ataxia and mental deficiency since early life. Postmortem examinations showed severe cerebellar granule cell loss. One child showed delayed motor development and mental deficiency in infancy. Other features included small head, cataracts, increased knee jerks, and intention tremor [Norman, 1940]. The disease locus was assigned on chromosome 9q34–qter [Delague et al., 2001]. It is not certain if SCAR2 is distinct from the disorder called cerebellar hypoplasia (OMIM 213000).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


