The EEG in Congenital Malformations of Cortical Development, Neurocutaneous Disorders, Cerebral Palsy, Autism/Mental Retardation, and ADHD/Learning Disabilities of Childhood
Mary Repole Andriola
This chapter reviews numerous disorders of infancy and childhood considered congenital with a known genetic or familial basis or acquired by insults to the developing brain. These include various structural abnormalities, cerebral palsy, autism, mental retardation, attention-deficit hyperactivity disorder (ADHD), and learning disabilities.
CONGENITAL MALFORMATIONS OF CORTICAL DEVELOPMENT
Anencephaly is a severe anomaly with absence of the cerebrum and superior skull. These infants survive only a very brief time; thus, EEG studies are not usually obtained for practical and technical reasons.
Hydranencephaly refers to nearly complete absence of the cerebral hemispheres that is thought most likely due to bilateral cerebrovascular occlusions or intrauterine infections with cytomegalovirus (CMV) or toxoplasmosis after the fourth month of gestation. The infant is often initially normocephalic and nondysmorphic. The EEG is usually attenuated (1) but Velasco et al. reported a hydrancephalic patient with seizures considered consistent with the Lennox Gastaut syndrome, though the typical slow spike-wave pattern was not seen (2).
Hydrocephalus refers to a dilatation of the ventricular system due to a variety of underlying brain structural anomalies or secondarily acquired from bleeding or infection. It is often treated by a neurosurgical procedure of shunting, typically with a catheter from the right lateral ventricle to the peritoneal cavity. The underlying abnormalities of brain structure, the presence of the shunt (the burr hole as a craniotomy), and at times complications of the shunt with infection, hemorrhage, and multiple revisions can give rise to EEG abnormalities and seizures. Ines and Markand (3) reviewed the EEGs of 92 hydrocephalic patients and found a higher incidence of EEG abnormalities in the shunted (95%) as compared to the nonshunted patients (47%), with a 65.4% versus 18.2% incidence of seizures in the two groups. Many of the EEG abnormalities localized to the area of the shunt itself and were first noted after surgery, though asynchrony of sleep spindles was noted in both groups (3). Bartoshesky et al. (4) found in studying a group of 111 children with meningomyelocele that 24 out of 98 children with shunted hydrocephalus had seizures. More of these children had other brain malformations besides Arnold Chiari, shunt infections, and perhaps shunt revisions and these children were more likely to be developmentally delayed than the rest (4). Saukkonen et al. (5) following 168 shunted hydrocephalic children found seizures in 22% of children prior to surgery in addition to abnormal EEGs. Some of these patients presented with seizures as newborns and some had infantile spasms with hypsarrhythmic EEGs but there was no specific localization or EEG pattern otherwise. The subsequent occurrence of seizures, 25.6% after shunting for a total of 47.6% over a long follow-up period with a mean of 8.9 years, they found related to the lack of use of prophylactic antiepileptic medication causing them to recommend treatment in those hydrocephalic children prior to shunting with abnormal EEGs with epileptiform discharges. They did not find a relationship of EEG abnormalities to the site of the shunt, number of revisions, nor infections, but they did find the development of seizures related to the occurrence of the slit ventricle syndrome (5). A similar incidence of seizures (48.5%) was reported by Noetzel and Blake (6) in 68 children with congenital hydrocephalus not associated with myelomeningocele. Brain malformations, developmental impairment, and spike, spike-wave, or slow-wave paroxysmal EEG activity at seizure onset focal or diffuse correlated with intractable seizures while age of shunting, revisions, or infections did not. The few children without mental retardation were able to come off their medication after 3 years seizure free (6). Seizures may also occur in these children as a symptom of shunt malfunction (7). Faillace and Canady described the various EEG findings in 9 out of 15 patients with seizure activity in the presence of shunt malfunction. These included diffuse dysfunction and focal spike activity (Fig. 16.1) (8).
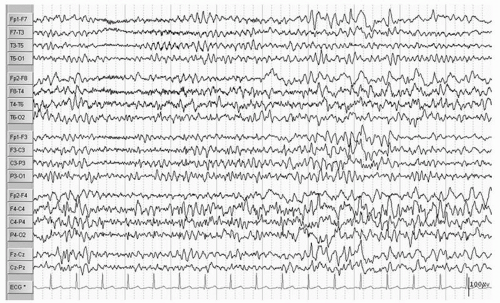 Figure 16.1 A 37-year-old woman with hydrocephalus, Arnold Chiari II, and myelodysplasia. Patient has had multiple shunts and revisions—initially on the right, now on the left. History of seizures as a child. EEG: slowing and higher voltage on the right plus independent sharp activity from either hemisphere. |
Holoprosencephaly (HPE) is a syndrome of congenital brain malformations with incomplete separation of the hemispheres and basal ganglia of varying severity often associated with midline facial dsymorphisms, hypothalamic pituitary dysfunction, developmental delay, abnormal EEGs, and epilepsy in approximately half the patients. A dorsal cyst may be present producing hydrocephalus that requires shunting. The disorder has multiple causes: teratogenic and chromosomal including trisomy 13 and 15 and the sonic hedgehog mutation (SHH), SIX, TGIG, and ZIC2. This can be considered an abnormality of neural induction in the ventrodorsal plane (9). There are four types: alobar, semilobar, lobar, and middle interhemispheric variant (10,11). Clegg et al. (12) reviewed the literature and reported on 32 children, approximately half of whom had epilepsy with various forms of the disorder. Of the 20 patients who had EEGs the abnormalities noted were hypersynchronous theta during waking and sleep, prominent spindle-like beta during drowsing and sleep, multiple epileptiform discharges, and periods of attenuation. Posterior gradient flattening was seen in one child with a dorsal cyst associated with alobar HPE (Figs. 16.2 and 16.3) (12).
Schizencephaly refers to a full-thickness gray matter-lined cleft of a hemisphere that may be unilateral or bilateral, open (communication between ventricle and subarachnoid space) or closed. The gray matter is abnormal, often composed of polymicrogyri or heterotopic gray matter. This congenital disorder of brain development occurring at the end of the first and the beginning of the second trimester may be associated with other cerebral anomalies, particularly agenesis of the corpus callosum, absence of the septum pellucidum, polymicrogyria, and gray matter heterotopias, neurologic and developmental impairment, epilepsy, and EEG abnormalities. Packard et al. (13) reported on 47 children with all types of which 57% had seizures, one third of which were difficult to control. The most common seizure type was complex partial (70%). Though seizures presented earlier and were harder to control in the bilateral open lip type, there was no correlation of seizure type or occurrence with the type of schizencephaly. Of the 27 patients with seizures, EEGs were unavailable in 1, normal in 3, contained spike and sharp wave discharges or sharp waves localizing to the cleft in 15, multifocal discharges in 5, and diffuse or focal slowing in 3. The neurodevelopmental outcome of these children was dependent on the extent of cortex involved. They often have motor and language impairment (13). The EEGs in nine patients with schizencephaly were reviewed by Granata et al. (14). Focal epileptiform discharges that correlated with their clinical seizures and the location of the cleft were the most common findings. Their six patients with unilateral clefts
had IQs in the normal range, hemiparesis, and incompletely controlled partial seizures that were postural at onset followed by turning or complex partial but did not generalize (Fig. 16.4) (14). A report of two cases by Iannetti et al. (15) raises the possibility of congenital CMV infection causing schizencephaly. These patients were negative for a mutation in the homeobox gene EMX2 that has been found in many patients with schizencephaly (15).
had IQs in the normal range, hemiparesis, and incompletely controlled partial seizures that were postural at onset followed by turning or complex partial but did not generalize (Fig. 16.4) (14). A report of two cases by Iannetti et al. (15) raises the possibility of congenital CMV infection causing schizencephaly. These patients were negative for a mutation in the homeobox gene EMX2 that has been found in many patients with schizencephaly (15).
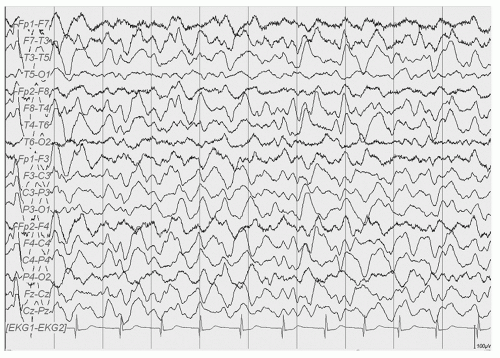 Figure 16.2 A 10-year-old girl with semilobar holoprosencephaly. The patient has marked developmental retardation and at times appears to have myoclonic spasms involving her upper trunk but her parents prefer not having her on medication. Her EEG is without age-appropriate waking and sleeping patterns and frequent high-voltage paroxysmal synchronous notched delta discharges as illustrated. |
Hemimegalencephaly refers to congenital hypertrophy of one cerebral hemisphere with abnormal chaotic cytoarchitecture of gray and white matter with an ipsilateral enlarged ventricle. Clinically, the patients have hemiparesis, mental retardation, and seizures. The EEG data in 12 patients reported by Paladin et al. (16) found partial seizures in all patients and 3 also had infantile spasms. In addition, they also reviewed the literature. Three EEG patterns were observed, tended not to
change over time, and were correlated with the patient’s clinical history:
change over time, and were correlated with the patient’s clinical history:
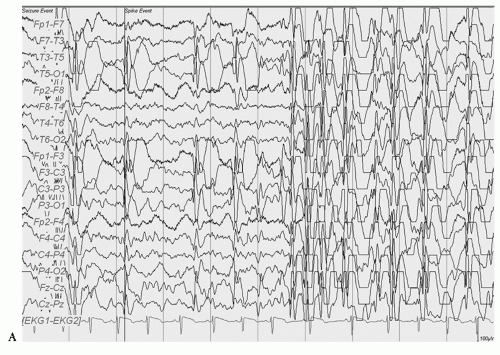 Figure 16.3 An 8-year-old girl with the middle interhemispheric variant of holoprosencephaly. She has mild facial dysmorphism, developmental delay, and symptomatic epilepsy. Her EEG reveals (A) asynchrony and multifocal spikes, (B) and marked activation by sleep consistent with ESES. (C) MRI demonstrates fused frontal lobes, absent septum pellucidum, and falx. (D) MRI shows agenesis of the body of the corpus callosum with radiation of the gyri. |
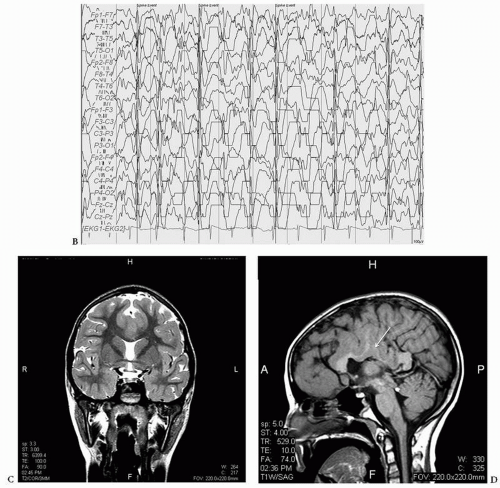 Figure 16.3 (continued) |
1. Triphasic spike complexes of large amplitude maximum in the abnormal hemisphere that appeared early as did seizures. No significant developmental milestones were achieved and one patient died at age 7 days.
2. Isolated “alpha-like” activity appeared at approximately 6 months of age in the central area and extended to the frontal by 2 years of age. Though seizure onset occurred before 6 months, these children learned to walk and talk and could attend special schools.
3. Suppression burst occurred at birth or within the first few months of life. This pattern seemed to respond to steroid therapy with a relatively favorable outcome in two patients (16).
Aicardi syndrome as first described in 1965 occurs in girls with severe mental retardation and ocular abnormalities who present with infantile spasms (17). Their brains have a thin unlayered cortex with diffuse polymicrogyria with fused molecular layer, nodular heterotopias in the periventricular region and centrum semiovale, and agenesis of the corpus callosum. The patients may also have chorioretinal lacunae, coloboma, and vertebral and costal abnormalities. They usually have a shortened life span. The EEG findings are that of completely asynchronous activity between the hemispheres consistent with agenesis of the corpus callosum plus multifocal discharges on a burst suppression pattern. There is also absence of recognizable stage II sleep activity such as spindles (18). There are more than 100 cases reported (19).
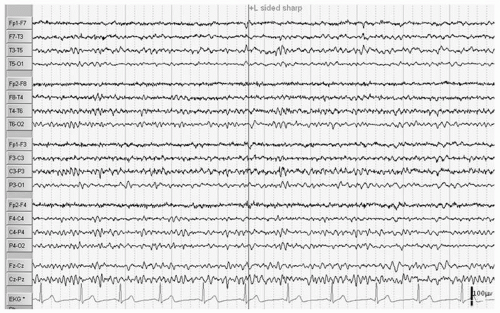 Figure 16.4 A 39-year-old man with incompletely controlled partial epilepsy and right hemiparesis secondary to left parietal open lip schizencephaly who nevertheless holds a job and is married. EEG findings of low-voltage left temporal sharp waves and underlying slowing. |
Agenesis of the corpus callosum may be seen as a total or partial phenomenon with the severity of clinical and EEG findings often related to other brain anomalies. Lacey (20) reviewed 40 children prospectively followed for up to 15 years. More than half of the patients were retarded and half had seizures. EEGs were done in all patients and abnormalities were consistent with the clinical picture from hypsarrhythmia in the seven patients with infantile spasms to background slowing and multifocal and generalized discharges in the patients with retardation and symptomatic epilepsy (Fig. 16.5) (20). Agenesis of the corpus callosum can also be caused by metabolic disorders (21).
When a lipoma occurs in the corpus callosum, epilepsy, usually partial and difficult to control, and an abnormal EEG, begins in childhood. The EEG abnormality may appear temporal or centrotemporal. Some of the patients are retarded or developmentally delayed. Gastaut et al. proposed that the seizures were related to the disconnection of the corpus callosum (22).
Intrauterine infections with viruses under the acronym TORCH (toxoplasmosis, rubella, CMV, and herpes simplex virus) plus syphilis can cause some of the congenital anomalies reviewed previously in this section. Infants with infections close to the time of delivery may have systemic symptoms. Sequelae include chorioretinitis, sensorineural hearing loss, hydrocephalus, microcephaly, developmental retardation, various neurologic deficits, and seizures with abnormal EEGs (23).
A number of teratogens are known to adversely affect the fetus. Fetal exposure to alcohol is a now-known preventable teratogen causing fetal alcohol syndrome (FAS) with microcephaly (as part of the intrauterine growth retardation), developmental retardation, and irritability and restlessness in infants. Craniofacial anomalies are also reported (24). A study by Kaneko et al. (25) comparing the EEG findings in 18 matched children with the FAS and Down syndrome versus normal controls using power spectra analysis found significant reductions in the mean power of alpha frequencies for both clinical groups. While Down syndrome children had diffuse slowing, FAS children had decreased posterior alpha especially on the left (25).
Another common modern teratogen inflicting harm on the fetal brain is cocaine. Infants exposed to cocaine have been
reported to have smaller head circumferences from birth through at least 2 years of age, and developmental delay when evaluated at 2 years (26). A report of 10 children with intrauterine exposure to cocaine and other street drugs found significant intracranial and ophthalmologic anomalies including absence of the corpus callosum, schizencephaly, hydrancephaly, porencephaly, septo-optic dysplasia, and optic atrophy (27). Half of the patients had seizures. Many of these malformations and their associated EEG findings have been described above.
reported to have smaller head circumferences from birth through at least 2 years of age, and developmental delay when evaluated at 2 years (26). A report of 10 children with intrauterine exposure to cocaine and other street drugs found significant intracranial and ophthalmologic anomalies including absence of the corpus callosum, schizencephaly, hydrancephaly, porencephaly, septo-optic dysplasia, and optic atrophy (27). Half of the patients had seizures. Many of these malformations and their associated EEG findings have been described above.
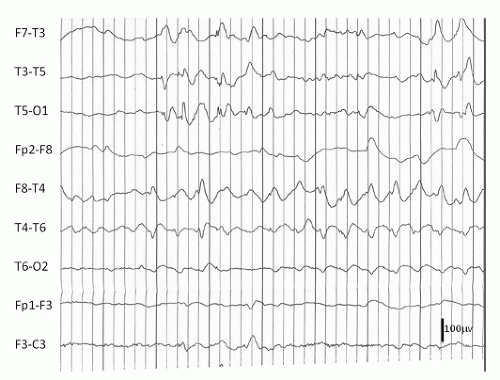 Figure 16.5 A 13-month-old female infant with agenesis of the corpus callosum, microcephaly, congenital cataracts, symptomatic intractable epilepsy, and severe developmental delay. The patient has a brother with the same disorder. Her EEG is markedly abnormal with lack of age-appropriate background rhythms, asynchrony, and multifocal spikes. Hemispherical asynchrony is the electrographic hallmark of this disorder. Her EEG at 5 months when she presented with infantile spasms had a hypsarrhythmic pattern. |
Congenital blindness may be associated with cerebral malformations, developmental retardation, or be purely on an ocular basis. The EEG of a blind child usually lacks occipital alpha while central rhythms are preserved. Occipital spikes may be seen in the absence of clinical seizures. These findings tend to decrease with age (28).
We should note, however, that the earlier theory that visual deprivation can cause electrical abnormalities over the visual cortex was not supported by some data, namely by the study of Jan and Wong (29) who investigated 104 children with ocular visual loss. In this study although most totally blind children’s records lacked alpha, five had spontaneously appearing and disappearing alpha rhythm.
A more recent study (30) showed a significant reduction of alpha power in the blind compared to the sighted controls over parieto-occipital recording sites and concluded that brain structures, associated with the generation of posterior alpha rhythms in sighted adults, might be either reorganized or atrophied following blindness from birth.
Congenital porencephaly is residua of an infarct that may have occurred during the pre- or perinatal period. The patient usually has a hemiparesis, developmental delay, and epilepsy. Ho et al. (31) describe the seizure localization in 14 patients with porencephaly and intractable seizures. The porencephaly was in the distribution of the middle cerebral artery in eight, posterior cerebral in three, internal carotid in one, and multiple vessels in two. The majority of patients had complex partial seizures. Scalp EEGs demonstrated abnormal background with focal slowing over the cyst in four patients. Eight patients had interictal discharges noted anterior temporally (one bilateral) and four extratemporally. Ictal monitoring confirmed temporal localization as did one intracranial monitoring. A total of 13 patients had hippocampal formation atrophy ipsilateral to the side of the lesion, 2 of whom underwent surgery and became seizure free (31).
Arachnoid cysts are intracranial congenital lesions in which redundant infoldings of the arachnoid membrane appears to trap cerebral spinal fluid into a cyst. These are often found “incidentally” when an imaging study is obtained post-trauma or as an evaluation for another diagnosis such as epilepsy. There has been a question raised as to whether or not these cysts play a causative role in a patient’s epilepsy and therefore should be treated surgically. The consensus appears to be that cyst removal is not likely to reduce seizures, but instead these patients need to be treated with medication even if epileptiform activity is noted in the region of the cyst (32,33).
Infantile nonprovoked epilepsy after the neonatal period is often related to cortical dysplasias. The pathogeneses and the pathology of malformations of cortical development are reviewed by Barkovich et al. (34). Focal cortical dysplasia refers to a localized area of abnormal cortical structure that may be appreciated now preoperatively by a combination of improved imaging and EEG studies. Patients tend to have intractable epilepsy with focal interictal discharges in about half the cases. They may be neurologically normal or impaired and may be candidates for seizure surgery.
Tassi et al. in a review of histologic sections from a large epilepsy surgery series correlated the clinical, EEG, MRI, and neuropath findings in patients with cortical dysplasia. They separate the pathology into the following three subtypes: (i) architectural dysplasia with disruption of cortical lamination and ectopic neurons in the white matter, (ii) cytoarchitectural dysplasia with abnormal cortical lamination plus giant neurofilament-enriched neurons, and (iii) (Taylor type) cortical dysplasia, abnormal cortical layering plus giant dysmorphic neurons, and balloon cells. Seizure frequency was more severe with the increased severity of the pathology. The epileptogenic zone was mainly extratemporal and the best seizure free rate after surgery was in the Taylor type (35). Krsek et al. (37) reviewing the results of 200 children who underwent seizure surgery for cortical dysplasia used the more complex classification system proposed in 2004 by Palmini et al. (36) that has more subgroups and correlated history, clinical findings, EEG, MRI, neuropsychological, surgical variables, and outcome data. Mild malformations of cortical development and focal cortical dysplasia type I were more frequently associated with perinatal risk factors. Ictal EEGs were more frequently precisely localized in the mild cortical dysplasia and the focal cortical dysplasia II group b (37). Kim et al. (38) report on 166 consecutive adult patients at the time of surgery
followed over a 10-year span. Forty additional patients were excluded as they also had hippocampal sclerosis. Scalp video EEG monitoring, interictal (42.7%), and ictal (76.5%) correctly localized to the resected lobe, while the MRI had a detection rate of 42.2% that was greatest the more severe the cortical dysplasia (38). For both series, the best surgical outcome was related to the completeness of the resection (37,38).
followed over a 10-year span. Forty additional patients were excluded as they also had hippocampal sclerosis. Scalp video EEG monitoring, interictal (42.7%), and ictal (76.5%) correctly localized to the resected lobe, while the MRI had a detection rate of 42.2% that was greatest the more severe the cortical dysplasia (38). For both series, the best surgical outcome was related to the completeness of the resection (37,38).
Congenital abnormalities of brain structure now known to be or likely to be on a chromosomal (genetic basis) such as neuronal migrational disorders often lead to EEG abnormalities and symptomatic epilepsy. Examples are periventricular nodular heterotopia (PNH), subcortical band heterotopia (SBH), and lissencephaly (34).
PNH are subependymal nodules of gray matter along the superior lateral walls of the lateral ventricles from the frontal horns to the trigones. Bilateral PNH occurs in females as an X-linked dominant (Xq28) usually with mild epilepsy onset in the second decade and normal to borderline IQ. The interictal EEG is normal or there are temporal discharges (Fig. 16.6). When the periventricular nodules are seen in association with other brain malformations, the patients usually are males, have a younger age at presentation, mental retardation, more difficult to control seizures (often atonic), and EEGs with focal and bisynchronous discharges (39).
Battaglia et al. (40) discuss the spectrum of PNH in 54 patients including the various types with known genetic point mutations of the FLNI gene in bilateral, symmetrical familial occurring mostly in females and familial patients with bilateral PNH not related to the gene and sporadic cases. Unilateral PNH is usually found in the posterior paratrigonal region of the lateral ventricle but can extend into white matter and cortical tissue. In this study, 12 out of 14 were females with 4 patients having abnormal overlying neocortical areas and 8 having ipsilateral hippocampal hypoplasia. Three patients were either autistic or had mild mental retardation (Fig. 16.7). Most patients had epilepsy with focal seizures difficult to control and the three who did not yet were under 15 years. Background rhythms were normal as were stage II sleep patterns. Photic driving was bilateral except in the unilateral cases where it was ipsilateral. Multifocal epileptiform activity was seen interictally even in the unilateral patients in addition to bilateral asynchronous abnormalities though in slow-wave sleep, bilaterally diffuse bursts of polyspikes were seen. Three ictal patterns are also described all partial onset. There is some evidence that seizure activity may arise from the heterotopic nodules (40,41). The patients may have hippocampal sclerosis; however, surgery to remove the hippocampus does not achieve seizure control (41).
Double cortex syndrome or SBH is characterized by a subcortical band of gray matter and an abnormal DCX gene (XLIS) in most cases (34). Mosaic mutations of the LISI gene that is usually the responsible gene in severe lissencephaly can be found in patients with posteriorly prominent SBH (42). These patients, more commonly female, are retarded with usually intractable epilepsy. The EEG pattern most typically seen is that of hypsarrhythmia with clinical infantile spasms followed by a Lennox Gastaut pattern though multifocal spikes may be noted (Figs. 16.8, 16.9 and 16.10) (43,44). In a 13-year-old female patient who had generalized tonic and grand mal seizures and the DCX gene mutation by DNA analysis, a persistent Lennox Gastaut slow spike-wave pattern was seen on EEG associated with SBH on MRI and increased PET FDG uptake in the band (45). Mutations of the X-linked gene DCX (XLIS) usually cause SBH in females and lissencephaly in males (46).
Related posts:
 Dynamics of EEGs as Signals of Neuronal Populations: Models and Theoretical Considerations
Activation Methods
Metabolic Disorders and EEG
Anticipating Seizures Based on EEG
Intracranial Monitoring: Depth, Subdural, and Foramen Ovale Electrodes
EEG Event-Related Desynchronization (ERD) and Event-Related Synchronization (ERS)
Dynamics of EEGs as Signals of Neuronal Populations: Models and Theoretical Considerations
Activation Methods
Metabolic Disorders and EEG
Anticipating Seizures Based on EEG
Intracranial Monitoring: Depth, Subdural, and Foramen Ovale Electrodes
EEG Event-Related Desynchronization (ERD) and Event-Related Synchronization (ERS)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


