(1)
Division of Critical Care Neurology, Mayo Clinic, Rochester, Minnesota, USA
Abstract
This chapter includes 18 clinical signs seen in acute neurologic conditions. The origin of the clinical sign and its first description is highlighted. A final comment provides an historical perspective.
Short Historical Note 1
Fixed Dilated Pupil
The Title of the Paper
Holman E, Scott WMJ. Significance of unilateral dilatation and fixation of pupil in severe skull injuries. JAMA. 1925;84:1329–32.

Fig. 3.1
Title page
The Paper and the Times
A “fixed pupil” in a comatose patient is an alarming sign, as everybody knows. It is such a staple of acute neurology that it will be hard to trace back the origin of the observation.
As early as the 1800s, abnormal pupillary reaction to light was noted in papers concerning traumatic brain injury, and one may assume most likely the first observation came with patients who had an acute subdural hematoma. One such description is by Richard Bright in his seminal textbook of Medicine co-written with Thomas Addison (Elements of the Practice of Medicine) in 1839. Bright noted a dilated pupil in a patient with epidural hematoma. The pupil became dilated on the second day after head injury. The patient became much less responsive and also developed a slow pulse. However, this clinical observation was not explained nor did any of the investigators that followed explain the association with compression or stretch of the third nerve.
Experimental studies already pointed out the importance of the dilated pupil. Ernst von Bergmann, a German surgeon, provided information in his 1880 classic book on head injuries. He mentioned fixed dilated pupils, but he was unable to clearly localize the lesion. In fact, von Bergmann—as many before him—believed strongly that the lesion was located in the cortex. In fact, his book has illustrations with possible cortical localizations of the oculomotor nerve [243]. Most noticeable was the experimental study by Leyden in 1866 who summarized the changes he found in increased intracranial pressure as follows: change in consciousness was followed by pupil enlargements at least temporarily, most often also in coma and often bilaterally, followed by circulatory symptoms that included abnormal pulse frequency, deep respiration, and later slow respiration. He clearly noted that with increased intracranial pressure, the pupils first became narrow, then dilated, but also noted that they were not always symmetric [245]. Other experiments followed and correlated pupil dilatation to increasing blood pressure waves and constriction with decrease. No further explanations were forthcoming.
Jonathan Hutchinson has been credited with describing, for the first time, a dilated pupil after a traumatic brain injury. In 1867, he documented a clinical pathologic correlation in two cases and noted that the third nerve was compressed, explaining the dilatation of the pupil [109].
Hutchinson described it in patients with “hemorrhage from the middle meningeal artery” and due to gravitation of blood toward the base. Jacobson later suggested it be named the Hutchinson’s pupil.
In 1887, however, Macewen provided more details, and his description is telling:
Many instances have been observed of dilatation and fixity of one pupil, while its fellow, perhaps with the exception of a little sluggishness of movement, remained normal. These were all cases of fracture of the middle fossa of the skull, and in comparatively few of them was the diagnosis verified by postmortem examination. … In those cases in which postmortem examination were obtained, the basal fracture was continued into or toward the vertex, and the clot in most instances occupied the whole of the middle fossa from the petrous portion of the temporal to the vertex, the convolutions, especially the ascending, being considerably compressed. … In those that recovered, the dilatation gave way slowly, and was followed by a degree of contraction, along with sluggishness of movement which ultimately passed off, though in one case it still remained small and sluggish at the termination of the fifth month from the date of the accident [144].
The fixed dilated pupil only became recognized as a key neurologic sign in the early twentieth century.
The Details of the Paper
Holman and Scott were perhaps the first researchers to point out the clinical significance of pupillary enlargement—the operation should be on the side of the fixed pupil (Fig. 3.1). They cited several previous observations ignoring this symptom and pointed out the paucity of descriptions of patients who deteriorated and lapsed into coma in the literature.
Holman and Scott found that anisocoria of the pupils may be transitory but that it was quickly followed by bilaterally widely dilated pupils. They suggested that “the mechanism of its appearance is not obvious, but it is assumed that the intracranial course of the third nerve, as it lies against the bony wall of the cranium, lends itself peculiarly well to compression from a pressure applied lateral and superior to it.”
The authors emphasized that “unilateral dilatation and fixation of the pupil is a valuable aid in determining the location of the intracranial injury and hemorrhage following head injuries.”
The Message and Acceptance
Papers followed that pointed toward the possible mechanism of a fixed pupil. These papers described experiments that noted stretch of the third nerve, compression against the clivus, and compression of the uncus of the temporal lobe, all possibly explaining how a third nerve palsy could mechanically occur [187, 227].
Holman and Scott’s observation was specifically cited by Rand who emphasized that an enlarged dilated pupil could indicate the site of the lesion [187]. This remained a fairly consistent finding when patients were examined. Rand concluded:
It’s believed that the hemorrhage is usually greatest on the side of the dilated pupil. … In cases in which hemiplegia and the dilated pupil were homolateral, intracranial hemorrhage was found on the same side. … In some cases one is unable to assign any cause for the homolateral hemiplegia and other extensive damage to the opposite cerebral hemisphere has been found at autopsy. This may account for the contralateral hemiplegia.
Reid and Cone, who performed animal experiments documenting the appearance of a dilated pupil in a monkey, eventually cemented the origin of fixed dilated pupils. They rapidly infused Ringers solution through a burr hole and found compressed oculomotor nerves from displaced hippocampal gyrus [189].
The clinical sign rapidly became mentioned in neurology and neurosurgery textbooks, and additional observations were added over time. Currently, we know that a unilateral fixed dilated pupil is seen early and can be followed by bilateral fixed pupils in a patient with an acute hemispheric mass. The shape of the pupil may change and become irregular, “football,” or oval-shaped. An oval-shaped pupil ipsilateral to the mass is most frequently seen but transitory, quickly becoming round, midsized, or dilated and fixed to light. Increased intracranial pressure is commonly associated with an oval-shaped pupil [154]. The shape may be explained by differences in parasympathetic tone in various segments and thus could imply midbrain involvement rather than peripheral fibers.
The presence of a unilaterally fixed pupil is now considered by many neurologists and neurosurgeons a result of an oculomotor lesion from direct compression, compression of the midbrain oculomotor complex, or traction of the oculomotor nerve against the clivus. There is surprisingly little proof of any of these theories, and it has been made clear that clinicopathologic correlation—due to time passed between both observations—is unreliable [193, 194].
Short Historical Note 2
Decerebrate Rigidity
The Title of the Paper
Bricolo A, Turazzi S, Alexandre A, Rizzuto N. Decerebrate rigidity in acute head injury. J Neurosurg. 1977;47:680–98.

Fig. 3.2
Title page
The Paper and the Times
Decerebrate rigidity—a term originally coined by Sir Charles Sherrington [16, 207, 208] is a classic neurologic sign. Sherrington famously described decerebrate rigidity in his transection experiments in cats and demonstrated the existence of a transverse plane at the level of the corpora mammillaria, red nucleus, and between anterior and posterior colliculi. Transection at that level would produce decerebrate rigidity. Rigidity disappeared when a transection was performed caudal to that plane at the level of the vestibular nuclei. When after a mesencephalic section the cerebellum was removed, the signs of decerebrate rigidity did not disappear.
The counterpart in man had been described in multiple earlier observations, but its meaning—in physiologic sense—remained elusive [114, 198, 247, 260]. The clinical features of decerebrate rigidity were a sudden yielding with stretching of the muscle (“clasp-knife” phenomenon) and invariable presence of clonus or subclonus tendon reflexes. Extensor rigidity with head retraction had been noted by clinicians in comatose patients and was mostly apparent in patients with cerebral hemorrhage extending into the ventricles [198].
Kinnier Wilson found decerebrate rigidity in cases of cerebral hemorrhages, meningitis, and intracranial tumors [260]. He described three groups: patients with decerebrate rigidity and tonic fits combined, patients with a decerebrate posture without tonic fits, and patients with tonic fits without persisting decerebrate posture.
Hughlings Jackson reported similar observations in patients with cerebellar tumors. He likened them to “tetanus-like seizures” and described patients with opisthotonus, extension of the lower extremities, and flexion of the upper extremities.
In 1923, Walshe noted the following:
The patient lay motionless and unconscious on her back with the head in a median position. There was no trace of head retraction. The arms lay across the chest semi-flexed at the elbows with the forearms slightly pronated in the wrist and digit flexed. The legs lay extended and abducted with the feet plantar flexed. There was spasticity of moderate degree in all four limbs, definitely more pronounced in the arms than the legs [248].
Walshe also noted that there was clear evidence of further progressive functions of the “vital medullary centers.”
Mollaret and Bertrand’s 1945 monograph is equally of interest, and the authors again emphasized that what is seen clinically may be different from what was observed in animal experiments [164].
In their 1966 classic text, Plum and Posner stated that decerebrate rigidity was seen in four clinical circumstances: massive and bilateral forebrain lesions, rostrocaudal deterioration, destructive or expanding posterior fossa lesions compressing the midbrain and rostral pons, severe metabolic disorders and intoxications depressing the diencephalon and forebrain [180].
Most observations were case series until a comprehensive description by Bricolo and associates. The aim of this manuscript was to report in more detail the postural patterns and their association with other neurologic signs and to assess its significance.
The Details of the Paper
This study includes 800 patients with severe head injuries admitted to the Department of Neurosurgery of Verona Hospital (Fig. 3.2). More than two-thirds of patients required mechanical ventilation. Decerebrate rigidity developed in 317 of 800 patients (40%) and mostly early after admission. The authors divided decerebrate rigidity into several types: full, unilateral, alternating, combined and mixed decerebrate rigidity, and decorticate rigidity (Fig. 3.3). Full decerebrate rigidity was observed in 35% of the patients. “The symptoms consisted of clenching of the jaw and extension of the four limbs, the upper limbs more than the lower; the arms were also adducted and internally rotated, the shoulders lifted, and the feet in plantar flexion.” Unilateral decerebrate rigidity was observed in 12% of patients “who developed extensor motor activity of the limbs on one side.” Only 7% of the patients showed decorticate rigidity, with “triple flexion of the upper limbs, adduced on the trunk, and clenched fists, while the lower limbs were hyperextended.” Twenty patients with severe head injuries developed a more complex posture, consisting of extensor rigidity on one side and a decorticate posture on the other. Only five patients showed mixed decerebrate rigidity described as “flexor rigidity of the lower limbs and hyperextension of the upper limbs.” The authors felt it was more commonly seen after a noxious stimulus and therefore could have been easily missed by the observer.
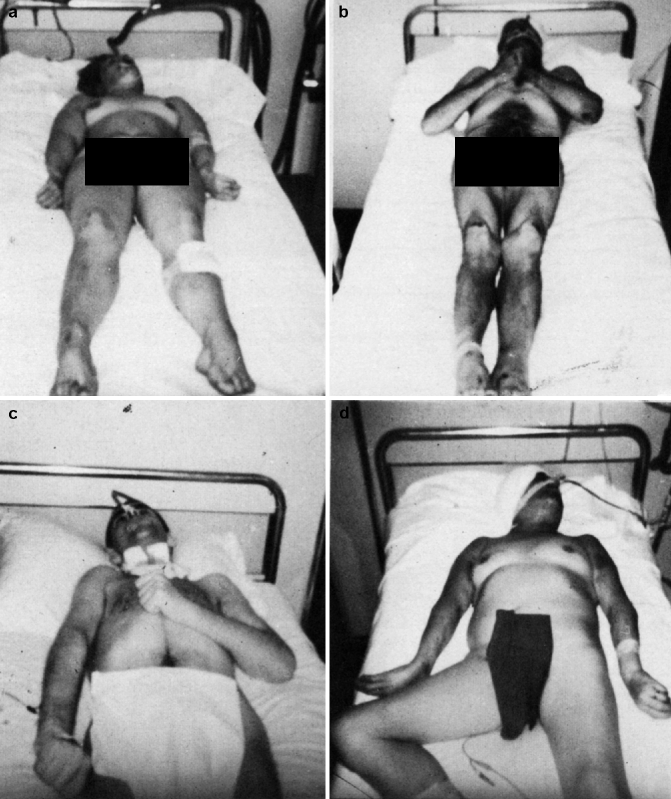
Fig. 3.3
Types of decerebrate rigidity (see text)
Most notable was the observation that the alternating decerebrate rigidity pattern comprised the largest group (40%), and the patients showed “a greatly variable postural attitude, either spontaneously or evoked, consisting of extensor or flexor rigidity in the same limb.” The authors also emphasized the common simultaneous occurrence of dysautonomia with cardiac arrhythmias, increased blood pressure, temperature disturbances, sweating, tachypnea, and periodic or irregular breathing. Mortality in patients with decerebrate rigidity increased to 80% and was twice as frequent than in patients without these motor responses.
The authors concluded:
The commonly accepted concept that in man decerebrate rigidity is the clinical expression of a physiological-anatomical result of brainstem transection must be rejected. … However, the most important finding from our data is that not all patients with decerebrate rigidity had structural lesions in the brainstem areas that were responsible for regulation of postural tonus. The absence of this close association in five cases provides further proof that, at least during the acute stage of head injury, decerebrate rigidity does not conclusively indicate brainstem structural damage. The localizing value of extensor motor abnormalities in patients with severe head injuries is not proven. … The use of terms such as “decerebration” and “decerebrate state,” because of their Sherringtonian implication, should be restricted to define a clinical condition due to severe midbrain dysfunction. This is implied when a stable and complete extensor posture combines with deep coma and brainstem ocular signs.
The Message and Acceptance
Decerebrate posturing has been observed in brain stem lesions and in lesions involving injury to both hemispheres without evidence of brainstem injury or tissue displacement. This motor response is often but not invariably an indicator of a severe structural brain injury [62, 129, 198, 261]. The site of the responsible lesion is both corticospinal and striospinal, and the response can be produced by injuries of all sorts. Precise localization of lesions on computed tomography or magnetic resonance imaging scans have not been possible nor has it been feasible to confirm the exact lesions as demonstrated by Sherrington in his animal experiments. These abnormal motor responses remain an important indication of the degree of brain injury and often are accompanied by other signs of brainstem injury. Decorticate and decerebrate posturing are likely a representation of a more diffuse lesion and can be part of a rostrocaudal deterioration pattern in patients with an expanding mass resulting in flexion, extension, and flaccidity, in that order. Decerebrate postering may persist and when it does-irrespective of its cause-portends a somber outlook. When it disappears recovery is quite likely.
Short Historical Note 3
Cushing Reflex
The Title of the Paper
Cushing H. Concerning a definite regulatory mechanism of the vasomotor centre which controls blood pressure during cerebral compression. Bull Johns Hopkins Hosp. 1901;12:290–2.

Fig. 3.4
Title page
The Paper and the Times
In 1783, Monro published “Observations on the Structure and the Function of the Nervous System.” Monro’s basic premise was that the skull was a rigid container filled with brain and blood. In 1846, however, Burrows added cerebrospinal fluid (CSF) to these components and also claimed that any change in any of these three components had to lead to a change in the other components for intracranial volume to remain constant.
At the turn of the nineteenth century, researchers became interested in the effect of increased intracranial pressure (ICP) as a result of increased intracranial volume. Von Bergmann was one of the early pioneers in explaining intracranial pressure following trauma [243, 244]. Von Bergmann’s experiments, using high pressure suddenly applied to the brain, described animals with “clonic spasms”. Slow heartbeat, deep and snoring respirations, vomiting, and incontinence were other remarkable symptoms. These symptoms were not seen when ICP was slowly increased. Immediately after injury, blood pressure rises and then falls. They interpreted apnea as a “paralysis of the respiratory center,” but if mechanical ventilation was applied, the pulse remained strong. Hill suggested that these early effects were due to diminished flow in the bulbar centers. Hill also concluded that death occurs when ICP equals the blood pressure in the carotid arteries [102].
Cushing’s physiologic contributions have become legendary, and his name became attached to this phenomenon, which was described in some detail by many before (Fig. 3.4). Increase in blood pressure with brain compression became known as “Cushing’s law” or “Cushing’s response,” or even “Cushing’s phenomenon,” but a better term is Cushing reflex [76].
The Details of the paper
The experimental setup was simple. Cushing used dogs, cats, and monkeys in his experiments [54–57]. The animals were anesthetized with ether. A catheter placed in the femoral artery recorded the blood pressure through a mercury manometer. He also recorded respiration and combined the records to show the changes over time. Subsequently, he made a large midline trephine opening, opened the dura, and placed a glass window (“fenster”) in the opening of the skull to expose the venous convolutions and pial arteries in order to observe changes in caliber (Fig. 3.5). Another burr hole was made over the cervical cord with opening of the dura. A metal cannula was screwed into the trephine hole to which an intracranial soft rubber tube was attached. The ICP was raised by filling the rubber bag with saline. Cushing observed blanching of the cortical arteries when the ICP increased and an improvement when blood pressure rose.
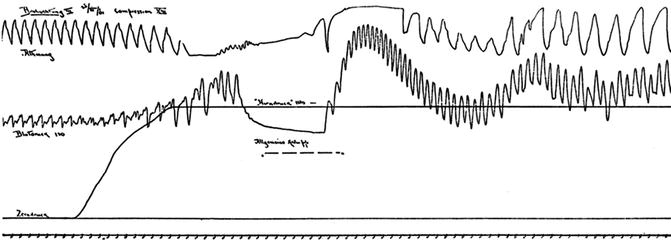
Fig. 3.5
Experimental setup (sketch from original article)
Cushing noted that until the ICP (“Hirndruck”) exceeds that of the blood pressure and the pressure rises slowly, only mild tachycardia and tachypnea are seen (Fig. 3.6). However, when the ICP rapidly rises, “Kussmaul–Tenner”spasms (extensor rigidity), bladder and bowel incontinence, apnea, and a “prominent vagus effect” meaning bradycardia and asystole were noted. Again the “vagus effects” could be avoided with a much slower rise in blood pressure. Cushing was able to repeat the experiments. “With reasonable limits of compression, however, this compensatory action may be indefinitively prolonged.”
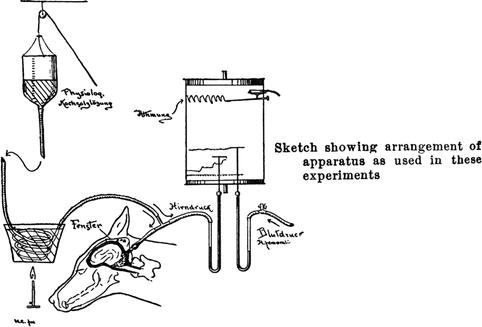
Fig. 3.6
Tracing of Cushing’s experiment. The figure shows increased blood pressure (Blutdruck), increased intracranial pressure (Hirndruck), absent breathing (Athmung), and spasms (Krämpfe)
Cushing summarized his results as follows:
As a result of these experiments a simple and definite law may be established, namely, that an increase of intracranial tension occasions a rise of blood pressure which tends to find a level slightly above that of the pressure exerted against the medulla. It is thus seen that there exists a regulatory mechanism on the part of the vasomotor centre which with great accuracy enables the blood pressure to remain at a point just sufficient to prevent the persistence of an anaemic condition of the bulb, demonstrating that the rise is a conservative act and not one such as is consequent upon a mere reflex sensory irritation.
The Message and Acceptance
Cushing’s data found its way into multiple publications. A major historical work on the subject found that the experiments on the vasopressor response were published in five communications in German and English between 1901 and 1903; four papers authored by Cushing and one chapter included in Kocher’s book on cerebral concussion [54–57, 76, 124, 125]. Kocher and Cushing advanced the field substantially by proposing the following stages of medullary compression:
Stage 1: Accommodation (kompensation). In this stage, the CSF is displaced out of the cranial vault followed by encroachment upon the cerebral venous bed with little change in the systemic circulation.
Stage 2: Early clinical manifestations (anfangsstadium des manifesten hirndruckes). In this stage, blood from the capillaries has been expelled; “anoxemia” of the vital bulbar centers results in rise of the systemic blood pressure. The pulse rate is retarded, but the pulse has a full quality. The respiratory rate also is reduced.
Stage 3: Advanced clinical manifestations (hohe stadium des manifesten hirndruckes). In this stage, the respirations are more snoring and rhythmic and may become Cheyne–Stokes type. Papilledema is seen, and pupils become irregular.
Stage 4: Medullary collapse (lahmungs stadium). In this stage, the “vital centers are exhausted.” The blood pressure is decreasing, and patient is in shock with all reflexes abolished, pupils dilated, and irregular respirations with apneic episodes.
In 1928, Heymans further refined Cushing’s findings by showing an initial tachycardia associated with hypertension just before the onset of bradycardia [101]. Others described a state of sympathoactivation secondary to medullary hypoxia mediated by depolarization of rostral ventrolateral medulla presynaptic neurons [89, 225, 253].
Changes in breathing patterns became an important cue for increased ICP. In a comparable experiment, Jennet found increased respiratory rate followed by irregularity, depression, and periods of apnea to be reproducible signs of increased ICP [115].
It is well known that many researchers before Cushing did very similar experiments. These included Paul Cramer, Ernst von Bergmann, Ernst von Leyden, Georg Althann, Friedrich Jolly, Friedrich Pagenstecher, Henri Duret, Bernard Naunyn, and Julius Schreiber. Nonetheless, Cushing’s experiments and data deduction have been considered far more careful, elaborate, and refined than his rivals [76].
Short Historical Note 4
False Localizing Signs
The Title of the Paper
Collier J. The false localizing signs of intracranial tumor. Brain. 1904;27:490–508.

Fig. 3.7
Title page
The Paper and the Times
Once clinical signs are known, they are traced to a specific location in the brain, and this approach has been axiomatic for both neurologists and neurosurgeons. The explosion of neuroimaging modalities may have challenged the clinical ability, and clinicians have become well aware that not infrequently magnetic resonance imaging scans point toward a different location or even explanation. However, early in the 1900s with a plain skull x-ray as the only imaging test, signs were recognized that could “falsely” point toward another region of the brain than predicted on the basis of anatomical knowledge. The most common “error” made by a neurosurgeon was operating on a presumed infratentorial tumor only to discover no tumor existed [71, 240]. The most commonly reported false localizing signs were lesions of the cranial nerves, particularly palsies of the abducens, trigeminal, and oculomotor nerves [11, 131].
Acute false localizing signs also were relatively common in the acute setting. Neuro-ophthalmologic signs were mostly encountered, such as a sixth nerve palsy associated with increased intracranial pressure and a fixed pupillary dilatation opposite from the actual site of the mass lesion [63, 92, 170, 192, 228]. Hemiparesis at the same site of a hemispheric lesion causing tissue shift also had been recognized but was more common in more slowly expanding tumors [117].
The Details of the Paper
Collier described that “false localizing signs” were mostly attributed to “indirect intracranial results of intracranial new growth” (Fig. 3.7). These signs were found in 13% of the 161 cases. The abducens nerve was affected more than any other cranial nerve, more commonly right-sided, and also equally unilateral or bilateral. The abducens lesion has been traditionally explained by the fact that it has a long course, but Collier suggested that the sixth nerve was the only cranial nerve that pursues a longitudinal rather than oblique or transverse course. Therefore, with backward shifting of the brain stem, the sixth nerve is easily damaged. In his words:
If the paralysis of these nerves is the result of shifting backwards of the brainstem from supratentorial pressure, the effect being direction upon the nerves attached to the brainstem, in proportion as that direction is more nearly frontal caudal direction, then the shifting backwards of the brainstem would cause paralysis of the sixth nerve first, then of the third nerve, and lastly the seventh and eighth, while the nerves which are transversely directed or with sinuous course would be little affected.
Collier also describes the occurrence of Jacksonian epilepsy, and general convulsions with tumors of the brainstem and cerebellum. These patients had significant ventricular distention but no other lesion that could explain the “local convulsion.”
Collier also considered vascular lesions in regions remote from the tumor that could give rise to false localizing signs and a misdiagnosis. Other explanations were locally spreading edema. However both causes were less clearly identified at autopsy.
The Message and Acceptance
Collier introduced the term “false localizing sign”, which remains the best description of the phenomenon. Others suggested terms, such as “nonlocalizing signs,” particularly for neuro-ophtalmology signs such as divergence paresis and convergency insufficiency with no clear precise localization [137].
The presence of an abducens paresis in supratentorial tumors fascinated many neurosurgeons. Stretching of the nerve due its long course or compression at its exit at the pontomedullary junction, compression by the anterior inferior cerebellar artery, and compression at the clivus exit were alternative explanations, but Collier rejected that notion. The presence of false localizing signs also interested Harvey Cushing. He suggested that abducens palsies may in some cases be attributed to arterial constriction and that also would explain the recovery.
The rarity of false localizing symptoms was noted in one study of 250 cases of intracranial meningioma with no evidence of palsy of the ninth, tenth, eleventh, and twelfth cranial nerves but with false localizing signs in 20 tentorial tumors [83]. Other single cases were reported, for example, a left parietal astrocytoma presenting with a fifth and eighth right cranial nerve palsy [68].
Most mystifying are patients with a fixed pupil opposite to the acute hemispheric lesion [41, 45, 155]. Traditional explanations have been compression of the oculomotor nerve by the herniating uncus of the temporal lobe (the pupiloconstrictor fibers are superficially located and easily injured) [226]. The opposite pupil becomes involved when the brain stem shifts downward, pulling on the opposite oculomotor nerve and causes bilateral fixed and dilated pupils. Thus, dilation of the opposite pupil at onset is hard to explain. Rotation of the brain stem in the axial plane could slacken the ipsilateral oculomotor and stretch the opposite oculomotor. Ischemia of the cranial nerve nucleus is another distinct possibility [45, 89, 195].
False localizing signs also have been described in spinal cord lesions with compressive cervical myelopathy causing a false localizing thoracic sensory level largely explained by ischemia of the medial portion of the spinothalamic tracts [171, 216]. Other false localizing signs pertaining to the practice of neurocritical care are pseudo-internuclear ophthalmoplegia—suggesting a brainstem lesion—in myasthenia gravis. Many other acute signs, such as an acute Horner’s syndrome in intracerebral hematoma have never been explained satisfactorily.
Short Historical Note 5
Brain Herniation
The Title of the Paper
Jefferson G. The tentorial pressure cone. Arch Neurol Psych. 1938;40:857–76.
McNealy DE, Plum F. Brainstem dysfunction with supratentorial mass lesions. Arch Neurol. 1962;7:10–32.

Fig. 3.8
Title page

Fig. 3.9
Title page
The Paper and the Times
In the early 1930s, most of the reports on neurologic findings in rapidly deteriorating patients involved expanding tumors. It was clearly appreciated at the time that tumors cause clinical signs as a result of invasion, compression, circulatory compromise, or as a result of abnormalities of the cerebrospinal fluid (CSF) circulation causing hydrocephalus. Shift with damage to brain tissue against the dural edge or over taut vessels also had been described.
When brains were examined at autopsy, pathologists noted that the uncus of the hippocampal gyrus was squeezed through the tentorial opening, and in the most severe cases with supratentorial lesions, the cerebellar tonsils had herniated into the foramen magnum. In 1920, Meyer was the first pathologist to describe herniation into the incisura tentorii (less known as Bichat’s cistern).
Despite suggesting that the lumbar puncture in the presence of herniation could be hazardous, the paper did not present any actual clinical examples.
In those days, lumbar puncture was considered a dangerous procedure for some patients, particularly in lesions of the posterior fossa. It was well known that this diagnostic test in patients with papilledema as a result of increased intracranial pressure (ICP), could cause further rapid worsening, often cessation of respiration. These concerns were addressed in Jefferson’s paper, and he described that herniation through the tentorium could cause compression of the brainstem and eventually could squeeze the contents of the posterior fossa through the foramen.
In the early part of the twentieth century, coma was described using several elements of neurologic examination but usually in combination with other physical findings. So, many textbooks would describe changes in breathing (slow, shallow, quiet, slow and noisy, sighing), pulse (weak, fluttering, rapid), odor of breath (urinous odor of uremia and acetone in diabetes), pupils (dilated, irregular, variable, contracted), and temperature (increased, high, subnormal). These were all unsuccessful attempts to grasp the difficulty of neurologic examination of coma. Perhaps as one of the first, DeJong’s 1950 textbook The Neurological Examination, included details on cranial nerve examination and emphasized the value of eye signs in localization.
In the early 1960s, more emphasis was placed on clinical findings that could help physicians recognize these acute deteriorations from displacement of tissue through the tentorium or through the foramen of magnum. A variety of acute clinical symptoms emerged—a fixed pupil, abnormal motor-responses, irregular or periodic breathing, and acute hypertension—and all of them were identified as indications for neurosurgical intervention [115, 116, 189, 238, 242].
What was unknown, however, was the precise clinical course and subsequent stages in patients who deteriorated. Many earlier publications describing a large variety of neurologic symptoms were difficult to summarize into a pattern and were not categorized in different stages [201, 226, 241]. This changed with the publication of McNealy and Plum’s paper. Their observations would introduce the term “rostrocaudal deterioration”, which for the first time presented what appeared to be a predictable succession of clinical signs when a patient would spiral down.
The Details of the Paper
Jefferson’s report includes several cases, and they are described in detail after a brief introduction on regional and anatomical relations and a brief historical overview on the herniation patterns known to neuropathologists.
Jefferson’s first case was a patient with a parietal astrocytoma who developed a hemiplegia over several years and died suddenly after a lumbar puncture. Within a few hours after CSF removal, the patient developed severe headache and became comatose. His neck and arms were notably rigid, and his legs were in “extreme extensor hypertonus.” He also had “decerebrate attacks” with rigidity of the neck and adduction of the arms. At autopsy, marked deformity of the midbrain was noted.
The second case was a patient with a frontal meningioma who deteriorated rapidly on admission and presented with dilation of pupils and papilledema. Again, markedly increased tone was found with hypertonus in both legs and extensor of plantar responses. This patient died in “spontaneous hyperthermia.” The increased tendon reflexes and the hypertonicity were found to be suggestive of a “tentorial pressure cone.”
The third case was a patient with a meningioma in the temporal fossa who died suddenly during surgery. Pathology showed that there was a “double pressure cone, one into the hiatus tentorii and the other into the foramen magnum.”
The fourth case involved a patient with a worsening characterized by bilateral pupillary dilatation, half flexed legs that were very rigid, and extreme neck rigidity suggestive of meningitis. A glioblastoma was found with the temporal lobe pushed into the edge of the incisura tentorii.
Jefferson suggested that the tentorial pressure cone could be recognized by the development of a “decerebrate state linked with pupillary alterations, such as the ‘hutchinsonian pupil’.” The descriptions, albeit new, lacked neither further neurologic detail nor a comprehensive explanation.
Several decades later, McNealy and Plum’s article appeared. The study included 52 patients with a supratentorial mass, who were seen over a 24-month period and who were selected from serial observations in 220 comatose patients admitted to the emergency room at King County Washington Hospital. The patients were selected for examinations when the lesions were inoperable or when they had surgery and continued to deteriorate. Neurologic examination was done by both authors at regular intervals up to 6 h. Clear notice was made of respirations, pupils, oculovestibular reflexes, and motor function. Respiration was observed for pattern and depth and brainstem reflexes, in particular the pupils, were categorized in large, mid-position, or small pupils. The provoked ocular movements were tabulated as hyperactive, dysconjugate, impaired, or absent. The motor responses were described as decorticate rigidity and decerebrate rigidity following noxious stimulation via the trigeminal (deep supraorbital pressure) and spinothalamic (skin pinch, testicular pressure, and Achilles tendon pressure) pathways. The authors were able to identify 27 cases with what they called “central syndrome” and 7 patients with what they called “uncal syndrome”. These syndromes progressed, according to the authors, in stages. The central syndrome (Fig. 3.10) progressed from a diencephalic stage, in which respiration was frequently interrupted by sighing or agitation. In this early stage small pupils (1–3 mm), easily elicitable horizontal doll’s eye movements, in addition to a unilateral hemiplegia reflecting the primary lesion, were noted. This then progressed to a midbrain upper pons stage in which Cheyne–Stokes respiration changed to sustained hyperventilation, small pupils changed to a fixed mid-position, and doll’s head eye movements became progressively more difficult to find. With further progression, a lower pontine upper medullary stage appeared and eventually, the patient progressed to a medullary stage in which the respiration became slow, irregular in rate or depth, and eventually interrupted by deep sighs or gasps. At this time, the pupils dilated widely and no stretch reflex could be obtained.
The manuscript also described in detail the development of third nerve midbrain stage in the setting of uncal herniation and a combination of both central and uncal herniation. The authors found that sudden progression, skipping over stages instead of a predictable rostrocaudal deterioration, was typically found in a patient with a cerebral hemorrhage that ruptured in the ventricle.
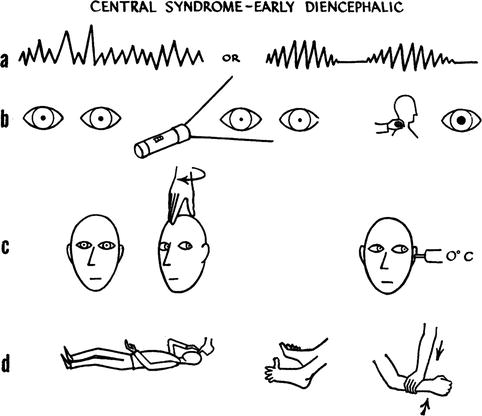
Fig. 3.10
Signs used to define a herniation stage by Plum and Posner (i.e., central syndrome)
The Message and Acceptance
For the first time, Jefferson’s paper further elucidated the clinical features of increased ICP and compression of the brainstem through herniation of brain tissue. The herniation could be either through the tentorial opening or through the foramen magnum. The clinical features involved sudden presentation of pupillary changes, neck stiffness, and extensor posturing. Hyperthermia often also was noted indicative of “central hyperthermia.” Jefferson referred to “Hutchinsonian pupil,” which was related to a unilateral fixed pupil associated with uncal herniation.
Clinical signs of tentorial and tonsillar herniation had been recognized by prominent neurosurgeons. Cushing also considered an increase in ICP to be the main reason for tonsillar herniation, and he felt that medullary compression would eventually cause respiratory failure. In a seminal case, a young boy developed respiratory arrest and Cushing decided not only to do supratentorial surgery but also to-albeit unsuccesfully-perform emergency suboccipital craniotomy to relieve the medullary compression believed to be caused by tonsillar herniation [206]. The author called it “a surgeon’s courage in times of despair” but more likely primary brainstem injury from displacement caused the signs rather than compression [206].
The central and tentorial herniation patterns, as described in this paper, became the basis of Plum and Posner’s monograph, “The Diagnosis of Stupor and Coma” (McNealy was one of Plum’s residents together with Swanson and Posner but McNealy died prematurely) [180].
For many decades since the original description, these patterns remained ingrained in neurologist’s minds and provided a teachable template. The didactics of neurologic examination of coma would now refer to these stages. However, the years that followed would find evidence to the contrary [152]. Very few patients—when they worsen—do progress in these stages with all four components—pupils, oculocephalics, motor response, and respiration—changing in a manner that can be exactly predicted.
Coning and herniation are well-accepted terms (and even a codable diagnosis), but the clinical correlates are less clearcut. Magnetic resonance imaging may show massive displacement but not with a perfect clinical correlate. The clinical entity of brain herniation—despite neuropathologic and radiologic findings—has not been fully worked out and in parts has remained quite inexplicable.
Short Historical Note 6
Neurogenic Hyperventilation
The Title of the Paper
Plum F, Swanson AG. Central neurogenic hyperventilation in men. Arch Neurol Psych. 1958;81:335–49.

Fig. 3.11
Title page
The Paper and the Times
Before respiratory patterns could be linked to specific brain injury, progress was made in the discovery of the morphology of the medullary respiratory center. Most of the early work can be attributed to Legallois’ experiments in 1812 in which stimulation of the pneumotaxic center increased the respiratory rate [136].
Abnormal breathing patterns had been recognized as indicative of a primary brain lesion and most commonly known were periodic breathing patterns. In the early papers on acute neurologic disease, the terms stertorous, gasping, or overventilation or hyperpneu were used frequently without further characterization.
There was reluctance to attribute hyperventilation to a brain lesion rather than to a compensatory response in acute illness. Rapid breathing was mostly seen as Kussmaul breathing in patients with a diabetic coma. Alveolar hyperventilation was typically observed as a response to hypoxia or metabolic acidosis or was associated with a specific pulmonary disorder. Hyperventilation with respiratory alkalosis also had been incidentally noted in hepatic encephalopathy and patients in the last stages of deep coma. These descriptions were followed by a series of studies linking brain injury to abnormalities of the rhythm of breathing. In the first half of the twentieth century, hyperventilation as a direct result from an acute brain lesion was not specifically mentioned in leading medical and neurologic textbooks.
The Details of the Paper
Hyperventilation associated with a brain lesion was first carefully described by Plum and Swanson (Fig. 3.11). The paper reports nine patients with acute lesions in the pons mostly caused by embolus to the basilar artery and compared them with 25 patients with brain lesions elsewhere who lacked hyperventilation (The control cases involved bilateral hemispheric lesions and unilateral hemispheric lesions.).
In their original description, the patients all had clinical signs pointing to a pontine lesion. Many patients had constricted pupils with skew deviation of the eyes or disconjugate motion, including internuclear ophthalmoplegia in four patients. Decerebrate rigidity was also noted.
As expected, the patients with central hyperventilation had a marked alkalosis (Fig. 3.12). The authors also reported frequent other respiratory abnormalities with six patients having Cheyne–Stoke type breathing and periodic breathing, usually preceding the central hyperventilation.
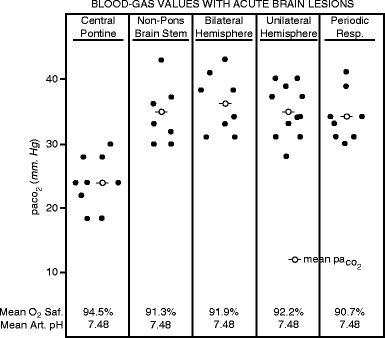
Fig. 3.12
Blood gas values in central neurogenic hyperventilation
The hyperventilation could be reduced or eliminated by oxygen therapy or by carbon dioxide inhalation. The study also carefully eliminated hypoxemia and metabolic acidosis as possible causes of hyperventilation. The authors also found that the arterial pH must fall to approximately 7.2 before respiration is stimulated by acidosis.
Autopsy demonstrated medial pontine destruction. The authors hypothesized that structures in the medial pontine reticular formation were inhibitory to respiration, and thus, a central neurogenic hyperventilation is the result of an uninhibited stimulation of the medullary respiratory centers by the lateral pontine reticular formation. The authors found only one report with disease in the cerebral hemispheres causing neurogenic hyperventilation.
The Message and Acceptance
It has been understood that both respiratory rate and alveolar ventilation are controlled by the respiratory centers in the pons and medulla. Thus, any neural, humoral input, or direct structural injury can lead not only to hyperventilation but also to hypoventilation or apnea.
Central neurogenic hyperventilation has now been well recognized in patients with catastrophic brain injury. Neurogenic hyperventilation can be seen in comatose patients with anoxic-ischemic encephalopathy and patients with upper brainstem compression and shift from a new hemispheric lesion [113, 234]. If neurogenic hyperventilation is present, the disorder is a consequence of a midbrain or pontine lesion and can be associated with pontine hemorrhages, embolus to the basilar artery, and progressive signs of brainstem compression from a hemispheric lesion [84, 100, 169, 234]. Later case reports found a correlation with brainstem tumors [84, 174, 209, 210]. Other signs localizing the lesion to the brainstem are often present. For unknown reasons, the incidence of primary brainstem lymphoma or astrocytoma in patients with central neurogenic hyperventilation is high, but it also has been reported in multiple sclerosis [231] and brainstem encephalitis [169], but again the responsible lesion is in the pons. The patient cannot inhibit respiratory drive; carbon dioxide rebreathing augments the patient’s need to breathe and increases the respiratory rate. Infiltration of tumor, whether lymphoma or astrocytoma, presumably destroys the inhibiting descending neurons from the pons to the medullary respiratory center. Lactate production from the tumor, another possibility, has been discounted by several studies as a major trigger.
In traumatic brain injury, periodic hyperventilation may occur with tachycardia, fever, and sweating and is related to sympathetic hyperactivity syndrome. It may be the most commonly underrecognized manifestation of acute brain injury and often the most commonly undertreated. Central neurogenic hyperventilation usually is continuously present and seemingly wearing out the patient. Hyperventilation in this setting also can be more erratic and may result in difficulty ventilating patients due to asynchrony with the ventilator. In sustained neurogenic hyperventilation, respiratory alkalosis is considerable with pH values greater than 7.6. This type of breathing disorder is best muted with infusion of potent respiratory depressants such as fentanyl.
Short Historical Note 7
Biot Breathing
The Title of the Paper
Biot MC. Étude Clinique et Expérimentale sur la Respiration de Cheyne-Stokes. New York: Harper & Brothers; 1878.

Fig. 3.13
Title page
The Paper and the Times
Several classic central periodic breathing patterns have been described. Biot breathing is a breathing pattern in patients with acute neurologic disease—also perhaps better known as ataxic breathing. Biot breathing can be contrasted with Cheyne–Stokes breathing (periodic stereotypical crescendo–decrescendo hyperpnoea followed by apnea) and apneustic breathing (periodic prolonged inspiratory hold). The breathing pattern Biot described is irregular and rapid, with intermittent pauses. Biot breathing is not commonly mentioned in the neurologic literature but surfaces occasionally in the anesthesia literature.
The Details of the Paper
Camille Biot made his seminal observations early in his career in the Hôtel Dieu Hospital in Lyon [22]. Biot wrote two main articles on breathing patterns. One large exploratory work focused on his observations on Cheyne–Stokes breathing [20, 21]. In each paper, he described patients who were admitted to the Hôtel Dieu Hospital with Cheyne–Stokes respiration, his major interest. In his first article, he described a patient who had respiratory movements that gradually decreased and increased but were irregular, and he was one of the first to publish recordings of this type of respiration in a 16-year-old patient with tuberculous meningitis [20].

Fig. 3.14
The first description of Biot’s breathing different from Cheyne–Stokes breathing
Translation: “We stated in the previous pages that the various authors who wrote about Cheyne-Stokes respiration had mentioned it in tuberculous meningitis. We have observed the case of a 16-year-old young man. We collected several pneumographic graphs. These graphs are conspicuously different from those that we saw in fig I, especially in that before and after the pause there is no respiratory movement that gradually decrease and increase; but, a respiration that is deep, dyspnoeic, like a big sigh from the patient during those moments. On the other hand Trousseau, which undoubtedly astute observation, noted in his Clinique (part II, page 240), that periodic irregularity of breathing is a sign of tuberculous meningitis; but the reading of this paragraph shows that it is not Cheyne-Stokes. Without wanting to come to a definitive conclusion, it seems that in meningitis it is not really the true type of Cheyne-Stokes respiration, but close to this type and more regular. It is an issue that should be the subject of future study.”
The breathing pattern Biot described is irregular and rapid, with rhythmical pauses lasting 10–30 seconds but sometimes with alternating periods of apnea and tachypnea. This breathing pattern lacked the crescendo–decrescendo cycles attributed to Cheyne–Stokes breathing and was completely irregular with varying periods of apnea. He named it rhythme méningitique.
In 1878, a larger thesis was published entitled Étude de Clinique et Expérimentale sur la Respiration de Cheyne–Stokes [21] (Fig. 3.13). This paper described theories of the origins of Cheyne–Stokes breathing. Biot argued that Cheyne–Stokes breathing included an increase in pulse during the pause and reduction of blood pressure during apnea. In this now discounted theory, the accumulation of carbon dioxide would increase the activity of the vasomotor center, and vasoconstriction would cause progressive ischemia of the respiration center resulting in deep breaths.
Biot added a section on earlier observations of the breathing pattern [14, 21] (Fig. 3.14 and 3.15). This respiration had been described as slow and with accelerating periods increasing to 10–20/min with unequal chest expansion: some short and incomplete, others long, deep, and often interrupted by gasps. The unevenness of breathing was not related to the heart rate. In another report, vomiting, constipation, and high fever along with short, incomplete, intermittent, and gasping breathing were considered characteristics of meningitis. He also recognized that similar patterns had been described in Trousseau’s textbook [239]. Sigismond Jaccoud thought this breathing pattern was an agonal phenomenon. In his description, the pulse slows down, and the breathing changes and becomes irregular and interrupted with deep sighs that are easy to recognize [112]. Biot concluded that a big sigh comes before the pause, and the periods are irregular. These were the two main characteristics that were always seen in meningitis and, thus, should be considered characteristic but different from the regular crescendo and decrescendo cycles of Cheyne–Stokes breathing. Biot concluded that this breathing pattern should be considered separately and not as a variant of Cheyne–Stokes breathing.
The Message and Acceptance
Many publications have inappropriately described Biot breathing as “periodic and consisting of groups of full respiratory effort” [5]. Biot breathing often is confused with cluster breathing, regular cycles of deep breaths with variable periodicity. An experimental study on Biot periodic breathing in cats created more confusion, and Biot breathing was called cluster breathing. Lesions in the pneumotaxic system of the rostral pons resulted in a breathing pattern that the authors explained as possible cyclic hypoxemia [252].
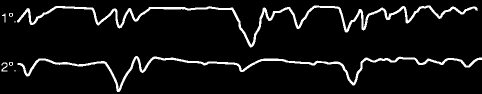
Fig. 3.15
The recorded periodic breathing pattern described by Biot
There is very little mention of Biot breathing in clinical papers, in particular those on meningitis. There are very few citations on clinical studies accurately documenting Biot breathing, confusing it again with cluster breathing. Plum and Posner, mentioned Biot in their description of ataxic breathing seen in the bulbar form of poliomyelitis [182]. It will remain unclear if Biot breathing had the same characteristics. Miller Fisher, in his work on neurologic examination of the comatose patient, correctly identified the main characteristics of Biot breathing and separated it from (regular) cluster breathing [74].
In the German literature, the eponym appeared first in Hofbauer’s 1904 book, Das Biotische Atmen, on the differential diagnosis of shortness of breath, but the author also stated he had not observed this type of breathing and in each case the breathing pattern was more likely Cheyne–Stokes breathing [105]. In the English literature, it appeared in the title of a 1911 review article by Connor, who observed seven patients, six of whom had meningitis [49]. Biot breathing may have become less obvious as a consequence of intensive care. One would expect that patients with this type of irregular breathing and poor oxygenation will likely be intubated as soon as they are seen and have their breathing assisted by mechanical ventilation.
Short Historical Note 8
Cheyne–Stokes Breathing
The Title of the Paper
Cheyne J. A case of apoplexy in which the fleshy part of the heart was converted into fat. Dublin Hosp Rep. 1818;2:216–23.

Fig. 3.16
Title page
The Paper and the Times
Of all “neurogenic” breathing patterns, Cheyne–Stokes breathing (CSR) is the most known but also most unrecognized. Not infrequently, physicians are called at the bedside for apneic periods only to discover it is part of a periodic breathing pattern. As early as antiquity, abnormal respiration has been noted in “apoplexy” and described often nonspecifically as infrequent and deep.
In the 1800s, studies appeared that periodically linked brain injury to abnormalities of the rhythm of breathing. Many physicians in the earlier days were able to describe abnormal breathing patterns, but it was John Cheyne and later William Stokes who described the ascending and descending waxing and waning pattern [175].
CSR often is related to changes in alertness and therefore should be part of assessing acutely ill neurologic patients [40]. Its appearance in a patient with a previously regular breathing pattern may be a valuable “neurologic” sign.
The Details of the Paper
Cheyne published his observation in Dublin Hospital Reports, a journal he had founded several years before (Fig. 3.16). In 1818, he described a 60-year-old man who was diagnosed with apoplexy and at autopsy was found to have a cerebral infarct. The patient also had a marked cardiomegaly and Cheyne found an “extremely irregular and unequal pulse.” His description is legendary:
The respiration is a first slow and heaving then irregular and sometimes convulsive and lastly, interrupted. The patient from relaxation of the palate, snores loudly during inspiration, and sometimes during expiration the upper lip, from relaxation or palsy is loudly blown up from the teeth, as we often see it upon great exhaustion, as in the subsiding of an epileptic fit. Sometimes the breathing is soft. Immediately before death the respiration is irregular and is performed perhaps not more than three of four times in the minute. the irritability of the heart survives the respiration sting with my finger over the artery of a person who died of apoplexy. I distinctly felt the pulse beat after the last expiration. Interrupted respiration is justly considered as the most dangerous symptom. … For several days his breathing was irregular; it would entirely cease for a quarter of a minute, then it would become perceptible, though very low, then by degrees it became heaving and quick, and then it would gradually cease again: this revolution in the state of his breathing occupied about a minute, during which there were about thirty acts of respiration [43].
Stokes felt the breathing pattern was pathognomonic, a fatty degeneration of the heart.
The symptom in question was observed by Dr. Cheyne, although he did not connect it with the specialties of the heart. … It consisted of the occurrence of a series of inspirations increasing to a maximum then declining in force and length, until a state of apparent apnea is established. In this condition, the patient may remain for such a length of time as to make his attendants believe that he is dead and a low inspiration followed by one more decided marks the commencement of a new ascending and then descending series of inspiration. This symptom as occurring in its highest degree I have only seen in the first weeks previous to the death of the patient [220].
Stokes’ recognition of this breathing pattern in congestive heart failure was equally important, and he recognized that its appearance had prognostic impact. This seminal observation is still true today despite marked improvement in medical management of congestive heart failure.
The Message and Acceptance
The periodic pattern was recognized but was frequently mentioned as a common symptom in CNS infections [53, 69, 175]. Multiple authors described the symptoms in tuberculous meningitis, often blurring the boundaries with Biot breathing. Others observed CSR with patients who had a hemorrhage into the medulla oblongata, linking it to an abnormality of the respiratory centers. The neurologic diseases that associated itself with CSR were meningitis, encephalitis, cerebral hemorrhage, cerebral infarcts, cerebellar hemorrhage, and ruptured intracranial aneurysm [175, 250]. Many authors recognized two separate types: one associated with intracranial lesions and the other with cardiac disease. Attempts to distinguish the two were many, but none were accepted. Hughlings Jackson, in 1895, emphasized the neurologic origin of Cheyne–Stokes breathing after he examined a patient deteriorating with a brain tumor: “The respiratory centers in cases of Cheyne-Stokes respiration due to a grave cerebral lesion will be left more than is normal to their own inherent automatism, and will presumably be more susceptible to the state of the blood, and perhaps to the influence of the vagi” [106]. In addition, he mentioned “In the cases of Cheyne–Stokes breathing the supposition I am considering is that higher level inhibition is taken off both medulla respiratory centers, whereupon, respiration proper being greater, apnea occurs, and the respiratory movements cease.”
Plum et al. hypothesized that CSR resulted from inappropriate periods of hyperventilation alternating with posthyperventilation apnea and found cortical dysfunction in all his patients, suggesting a preferential neurogenic cause.
CSR most often is seen in sleep. Later studies found that apnea duration bears no relation to circulation time or cardiac output [44, 119, 160, 167]. Current views include instability of respiratory control due to hyperventilation, prolonged circulation time, and reduced blood gas buffering capacity. More recent studies in patients with cardiac disease and stroke are confirming strikingly high prevalence of central sleep apnea with respiration. In central sleep apnea, there are cyclic apneas caused by transient lack of respiratory effort and not upper airway obstruction [104, 262]. CSR is often associated with impairments in sleep and are more common in patients with heart disease. Nonetheless, continuous positive airway pressure ventilation improved CSR in only 50%.
Miller Fisher’s observations in comatose patients are also noteworthy [74]. He identified several variations of CSR. There may be only waxing and waning without an apneic period. The respiration may be slow or rapid, and a combination of CSR and hyperpnea may occur. Miller Fisher also observed that high fever tends to abolish CSR. A short-cycle CSR with short apneic and hyperpneic phases of only a few breaths was seen in neurologic patients with intracranial disease. He eloquently concluded “when the comatose patient is breathing quietly, regularly and slowly as in sleep one usually can be sure that the neurologic state is not desperate” [74].
Short Historical Note 9
Ondine’s Curse
The Title of the Paper
Severinghaus JW, Mitchell RA. Ondine’s curse – failure of respiratory center automaticity while awake. J Clin Res. 1962;10:122.

Fig. 3.17
Title page
The Paper and the Times
There has been significant interest in discovering breathing patterns that could be highly specific for neurologic disease, naming them and setting them apart from the classic Cheyne–Stokes breathing pattern. Bulbar poliomyelitis had been traditionally associated with hypoventilation, but the mechanism here was different. Before applying the term Ondine’s curse to central hypoventilation by Severinghaus and Mitchell, a single case was described by Ratto et al with loss of automaticity of breathing after a brainstem stroke [188].
Severinghaus and Mitchell used a clever name for primary alveolar hypoventilation syndrome. Ondine’s curse now refers to patients with long episodes of apnea while asleep but who can breathe on command. They referred to the original German fable “the sleep of Ondine” by Giraudoux. The curse was a punishment for unfaithfulness and Ondine took away her husband automatic functions. It included loss of all automatic functions—seeing, hearing, moving, and breathing.
The Details of the Paper
The paper that coined the term was an abstract with little details (Fig. 3.17). Three patients with apnea after high cervical cord or brainstem surgery and may have had bilateral spinothalamic tract cordotomies for pain relief. In cervical tractomy for intractable pain, the reticulospinal tracts activate phrenic nerve motor neurons and ascending spinoreticular fibers.
The Message and Acceptance
The term Ondine’s curse has become entrenched in medical literature, but it remains a very rare form of sleep apnea [223]. The term has been used freely and not entirely correctly. It is now used for sleep apnea, hypoventilation syndrome, and loss of automatic ventilation [97]. Pulmonary pathology should be excluded, and typically, there is a rise in partial pressure of carbon dioxide with no response in ventilation. The condition can be mimicked by high doses of opioids. It has been described in congenital central hypoventilation associated with mutation of the PHOX2B gene, but in this condition, there is also a defect in cardiac control [46]. Acquired forms of Ondine’s curse always have involved lesions in the respiratory centers of the medulla oblongata. Multiple cases of central hypoventilation have been described with acute stroke, tumors, infections, multiple sclerosis, and cervical cordotomy [6, 25, 46, 47, 85, 151, 157]. Lesions are bilateral in the ventrolateral brainstem between the trigeminal nerve nucleus and the upper cervical level [218]. The disorder is of interest to critical care physicians but will likely be rarely diagnosed in its pure form and may be difficult to demonstrate [221]. Diaphragmatic pacing has been successful.
Short Historical Note 10
Asterixis
The Title of the Paper
Adams RD, Foley JM. The neurologic disorder associated with liver disease. Res Proc Assoc Nerve Ment Dis. 1953;32:198–237.

Fig. 3.18
Title page
The Paper and the Times
The early literature on coma specifically concentrated on how organ failure affects the brain. Well known are the descriptions of British physician Richard Bright leading to the triad “arterial hypertension,” “albuminuric retinitis,” and “manifestations involving the central nervous system.” Bright noted that patients in the earlier stages of renal failure developed “myoclonus” and could evolve into a more serious state with seizures.
Another challenge for physicians was to distinguish a set of clinical signs that would be characteristic for liver failure. The “mental” changes were often nonspecifically described as stupor. Frerichs’ description of liver disease involved two stages: excitement (delirium and convulsions) and depression (progressive stupor and coma). Frerichs emphasized a transitional phase of “gloomy, irritable temper and restlessness” but also “quiet, harmless wandering” [80]. The emergence of jaundice marked the development of delirium, convulsions, and coma.
Abnormal motor movements were noted, but the descriptions were far from distinct and without understanding of its physiology. Descriptions often included “muscular tremors,” “rigors,” “partial twitching or convulsions,” or “jactitations.” Others observed “choreiform movements” or “tonic spasms.”
Parallel to better understanding of these clinical manifestations were the neuropathologic descriptions of the consequences of the organ disease. Earlier observations included enlargement of nuclei of the protoplasmic astrocytes with similarities to Wilson’s hepatolenticular degenerations [1–3]. Adams and Foley would publish a major treatise that would “define more precisely the clinical neurologic syndrome presented by patients with different types of liver disease.”
The Details of the Paper
The material came from observations made by the neurologic and medical services and the Mallory Institute of Pathology of the Boston City Hospital and data obtained through assistance of collaborators from Massachusetts General Hospital (Fig. 3.18). Clinical observations were made on 60 patients who developed hepatic coma most from “Laennec’s cirrhosis of alcoholic type.” All patients admitted to the wards were examined carefully by the authors, and after an initial comprehensive neurologic examination, patients were examined daily after the first signs of hepatic coma. Special attention was given to testing of concentration (speed, subtraction) and the presence of abnormal movements. Adams and Foley noted “a peculiar intermittency of sustained muscle contraction that presents as a irregular flapping movement when the arm and legs are held outstretched, a fluctuating rigidity of the limbs grimacing, sucking and grasp reflexes and at times convulsions completing the clinical picture.”
Adams and Foley described it even more clearly in the following observation and confirmed using an oscillographic record of the outstretched arm that showed a brief succession of two to five beats of rhythmic discharge with a frequency of 8–16 seconds followed by a silent interval of 0.2–0.5 seconds that correspond with a lapse of posture (Fig. 3.19):
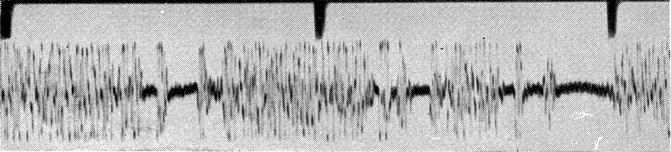
Fig. 3.19
Recording of asterixis
It is most consistently and most effectively demonstrated by having the patient hold his arms and hands outstretched with the fingers spread apart. Depending on the severity of the process, as this posture was maintained there appeared at irregular intervals of a fraction of a second to seven seconds, a series of movements consisting usually of lateral deviations of the fingers, flexion-extension of the fingers at the metacarpophalangeal joint and flexion-extension at the wrist. The movements were rapid and arrhythmic and one phase, i.e flexion when the arms were outstretched in a pronated position, was always more rapid that the other. They tended to occur in bursts, at a rate of one every second or two. … In the legs the abnormality of movement would be demonstrated most easily by having the recumbent patient elevate his leg and dorsiflex his foot. Then, at irregular intervals there would be a sudden flexion at ankle knee or hip followed by a slower extension. In the face strong closure of the eyelids strong retraction of the corners of the mouth or pursing of the lips were required to demonstrate this peculiarity of movement. It was also seen in the protruded tongue.
Adams also described for the first time a “paratonic rigidity” of the plastic type persisting throughout both passive flexion and extension of any joint. Repeated grimacing, together with motor restlessness, also was noted to be common.
Adams and Foley also described “a blunt spike and wave” activity that later was named “triphasic wave” by Bickford and Butt [19].
The Message and Acceptance
The term asterixis was not mentioned in the paper or in the attached discussion that concentrated on the neuropathologist findings. According to Laureno’s biography of Raymond Adams, the term was coined by Foley [132] Adams recalled that Foley, over a bottle of Metaxa in a Greek café, discussed a possible term with a classic scholar from Boston College, and they were searching for a word that indicated lack of steadiness. The term asterixis denoted inability to maintain posture (a-privative, sterixis-support).
Asterixis was considered “one of the most characteristic features of hepatic coma.” However, Adams and Foley pointed out in their original paper that the sign had been seen in uremia, polycythemia, and heart failure and hypokalemic stupors.
Other causes for asterixis would be reported over the ensuing years, and the sign is now part of the neurologist’s catalogue of clinical signs. Terms such as negative myoclonus seem confusing and have been discouraged [39]. A very good video clip of asterixis recently has been published [162].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


