The Localization of Lesions Causing Coma
Most causes of coma speedily threaten life or recovery of neurological function. Thus, they must be promptly identified and treated. Unfortunately, patients with a depressed level of alertness cannot give an account of the events leading to their situation, and often no one who has observed the patient before admission is available to provide such information. Thus, the physician has to rely on examination of the patient, not only to localize the damaged anatomic structures but also to identify the offending agent. The examination should be thoughtful and well informed but not necessarily long. A delay in protecting the airway of a poorly responsive patient may cause irreparable neurological damage [123].
The diagnosis of impaired alertness and coma is well reviewed in some excellent monographs [118,189]. The following pages draw heavily from these sources. Unlike in previous editions of this book, the diagnosis of death based on neurological criteria (brain death) is discussed at the end of this chapter. Despite the availability of neurophysiological and neuroimaging tools to help make this diagnosis, the neurological examination, object of this chapter, remains critical [178].
The Unresponsive Patient
Terms such as coma, stupor, lethargy, and the like indicate a depressed level of alertness. These terms, however, fail to convey vital information needed for neurologic localization and management. Rather than using one of these terms, a description of the patient’s level of responsiveness (incorporating some detail of the patient’s responses to diverse reproducible stimuli) facilitates communication among members of the health care team, enhances consistency in successive evaluations of the patient, and sets the basis for a rational diagnostic assessment. For instance, stating that “the patient was stuporous” provides little information. Instead, the real situation can be much better conveyed by explaining in everyday English that “Mr. Z lay motionless in bed unless called loudly by name, when he opened his eyes briefly and looked to the left. He failed to answer any questions or to follow instructions.”
Two terms have gained acceptance among neurologists and are widely used: akinetic mutism and locked-in syndrome. Akinetic mutism [141] refers to a state in which the patient, although seemingly awake remains silent and motionless. Only the eyes dart in the direction of moving objects, such as the examiner approaching the patient’s bed. The examiner, attempting to converse with a patient in this state, gets the distinct impression of failing to draw the patient’s attention and interest. Despite the lack of movement, there are few signs indicative of damage to the descending motor pathways. Instead, “frontal release signs,” such as grasp or sucking, may be present. Patients who remain completely motionless are not seen as often as those who move one side or one arm in a stereotyped fashion but in every other respect fit into the syndrome of akinetic mutism. In such cases, the paralyzed side may display signs of corticospinal tract involvement, such as hyperreflexia and a Babinski sign.
If a history is available, akinetic mutism can usually be distinguished from psychogenic (often catatonic) unresponsiveness. Otherwise, the diagnosis may be difficult. Particularly when exposed to painful stimuli (such as those caused by soiled linen or a decubitus ulcer) or to infection, patients with akinetic mutism appear excited and tachycardic and perspire heavily, thus superficially resembling a catatonic patient. Signs of frontal release or corticospinal tract damage favor the diagnosis of akinetic mutism. In the catatonic patient, the electroencephalogram (EEG) is normal (often desynchronized, with low-voltage fast activity), but in the patient with akinetic mutism, the EEG may show slow-wave abnormalities [102].
Lesions that cause akinetic mutism affect bilaterally the frontal region (anterior cingulate gyri), the diencephalo-mesencephalic reticular formation, the globus pallidus, or the hypothalamus [31,92,101]. Common causes are anoxia, head trauma, cerebral infarction, severe acute hydrocephalus, and direct compression by tumors [95,172]. Other extensive lesions, such as air embolism or end-stage degenerative or infectious brain disorders, such as Creutzfeldt–Jakob disease, can also cause this syndrome [3,51,109,150,175]. Metabolic or ictal disorders disrupting the same areas may give rise to a transient disorder of alertness similar to akinetic mutism [7,143]. A syndrome of transient mutism and relative akinesia may occur a few days after midline cerebellar or fourth ventricular surgery [18,21,36]. One such patient improved after dopaminergic stimulation, suggesting involvement of the dopaminergic pathways, at least in some cases [18]. Hyperkinetic mutism, with continuous bilateral ballism and dystonia, has been described in a diabetic with multiple subcortical and cortical infarcts [53].
When the cerebral hemispheres have sustained severe and widespread damage (such as that due to severe trauma, anoxia, or encephalitis), the patient may, after some weeks of complete unresponsiveness, evolve into a situation similar to akinetic mutism, with the return of sleep–wake cycles. These patients, however, demonstrate obvious signs of pronounced bilateral corticospinal tract damage. This situation, in which the patient’s functions are restricted to the autonomic sphere, has been termed the vegetative state [4]. The vegetative state has been defined as a chronic neurologic condition characterized by lack of awareness of self and external stimuli, accompanied by sleep–wake cycles, with preservation of vital vegetative functions, such as cardiac function, respiration, and maintenance of blood pressure. Patients in a vegetative state show no evidence of sustained, reproducible, purposeful, or voluntary behavioral responses to visual, auditory, tactile, or noxious stimuli; show no evidence of language comprehension or expression; have bowel and bladder incontinence; and have variably preserved cranial nerve and spinal reflexes. Persistent vegetative state is defined as a vegetative state present one month after acute traumatic or nontraumatic brain injury or lasting for at least one month in patients with degenerative or metabolic disorders or developmental malformations [4].
In addition to its pejorative connotation, the term persistent vegetative state assumes that it is possible from the physical examination combined with available clinical information to define accurately the patient’s state of awareness. Given that motor responses on the part of the patient are needed to evaluate the presence of awareness of self and external stimuli, in many of these patients with severe damage to motor mechanisms it is very difficult to assess the degree of awareness. The presence of extensive damage on MRI or CT is helpful but does not define this issue either [181]. In some patients meeting diagnostic criteria for the chronic vegetative state, functional neuroimaging has documented awareness and the ability to follow commands [97]. Given the shortcomings of the term persistent vegetative state, it is better to refer to this state as persistent unresponsiveness. Responses are all that the examiner can evaluate. Persistent unresponsiveness is a descriptive term reflecting accurately the information on the patient, and avoids the pitfall of making inaccurate assumptions as to the degree of awareness or the level of brain function in a given patient.
Recovery of consciousness from a posttraumatic persistent unresponsive state is unlikely after 12 months in adults and children [91]. Recovery from a nontraumatic persistent unresponsive state after three months is rare in both adults and children, but it can happen, usually with residual severe disability [9,37].
Even more vague and inaccurate than the term “vegetative state” is the more recent “minimally conscious state.” If a short description is to be used, “minimally responsive” seems much more appropriate [10]. Recovery from the minimally responsive state has been documented even after 19 years from onset [174].
The locked-in syndrome refers to a condition in which the patient is mute and motionless (deefferented) but remains awake, alert, aware of self, and capable of perceiving sensory stimuli. Although horizontal eye movements are often impaired due to involvement of the paramedian pontine reticular formation, the patient’s level of alertness can be gleaned from her response to commands involving vertical eye movements or eyelid movements. The EEG reflects the patient’s state of wakefulness. The locked-in syndrome is usually due to basilar artery thrombosis with ventral pontine infarction, pontine hemorrhage or tumor, or central pontine myelinolysis (osmotic demyelination syndrome) [50,96]. These lesions involve the descending motor pathways bilaterally in the basis pontis but spare the more dorsal reticular formation). Bilateral ventral midbrain lesions [23,41,94], tentorial herniation [68,183], Guillain–Barré syndrome [8,124], or myasthenia gravis may rarely cause this syndrome. In locked-in syndrome due to mesencephalic lesions, bilateral ptosis and vertical (as well as horizontal) ophthalmoplegia are present. Fou rire prodromique (pathologic laughter at the onset of a stroke) may rarely herald the onset of a bilateral ventral pontine stroke leading to a locked-in syndrome [176].
Anatomic Substrate of Alertness
In general, the maintenance of consciousness depends on interaction between the ascending reticular activating system (ARAS) and the cerebral hemispheres. Damage to the ARAS, described in animals by Moruzzi and Magoun in 1949 [99], induces a state of coma in which the animal becomes unresponsive and its EEG shows sleep patterns despite vigorous sensory stimulation. In humans, the ARAS lies in the paramedian tegmental region of the posterior portion of pons and midbrain [110]. It is a complex polysynaptic fiber system that extends from the superior half of the pons through the midbrain to the posterior portion of the hypothalamus and to the thalamic reticular formation (Fig. 23.1). The thalamus is the source of diffuse thalamocortical projections that regulate and coordinate cortical activity [58,84]. Sedative drugs act, at least in part, by interfering with the synaptic network of the ARAS, which is played on by sensory stimuli.
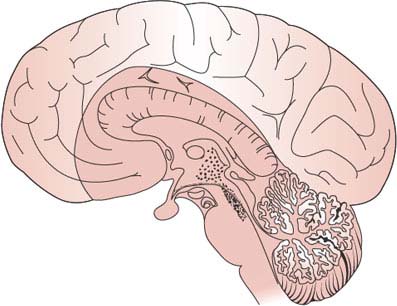
FIG. 23.1. Ascending reticular activating system (ARAS). The dotted area in this midsagittal section of the brain corresponds to the approximate location of the ARAS in the upper brainstem and the diencephalon.
The medial longitudinal fasciculi, which connect the abducens and oculomotor nuclei, and the oculomotor and trochlear nuclei themselves are situated amid the neurons of the pontine and midbrain portions of the ARAS. Thus, when unresponsiveness is caused by brainstem damage, the lesion affects the mechanisms of ocular motility as well, and its location can often be determined by abnormal patterns of ocular motility.
Bilateral cerebral hemispheric lesions may cause transient coma, particularly when they involve the mesial frontal region. Large unilateral lesions of the dominant hemisphere may occasionally cause transient unresponsiveness, even in the absence of a mass effect [1].
In the diencephalon, posterior hypothalamic lesions induce prolonged hypersomnia. Acute bilateral damage of the paraventricular thalamic nuclei is attended by transient unresponsiveness, followed, when the lesions are large, by severe amnestic dementia (see Chap. 18).
Signs with Localizing Value in Coma
In a comatose patient, the respiratory pattern, pupillary response, eye movements, and position or movements of the limbs provide important clues to the anatomic site and nature of the injury.
Respiratory Patterns
Although the respiratory pattern of a patient in coma may be helpful in localizing the level of structural dysfunction in the neuraxis [156], metabolic abnormalities may affect the respiratory centers of the pons (pneumotaxic and apneustic) and medulla (expiratory and inspiratory) and result in patterns resembling those due to neurologic disease (Fig. 23.2). Thus, caution and a thorough evaluation of the metabolic status of the patient must guide the interpretation of respiratory changes.
POSTHYPERVENTILATION APNEA
This condition reflects mild bilateral hemispheric dysfunction. Because demonstration of this respiratory abnormality requires the patient’s active cooperation, this sign is mentioned here mainly to clarify the genesis of other respiratory patterns. To elicit this phenomenon, the patient is simply asked to take five deep breaths. This maneuver normally decreases arterial pCO2 by about 10 mm Hg and, in the healthy patient, is followed by a very brief period of apnea (<10 seconds). The stimulus for rhythmic breathing when the pCO2 is lowered probably originates in forebrain structures, because sleep, obtundation, or bilateral hemispheric dysfunction abolishes it. Thus, when bilateral hemispheric lesions are present, the posthyperventilation apnea lasts for as long as 20 or 30 seconds.
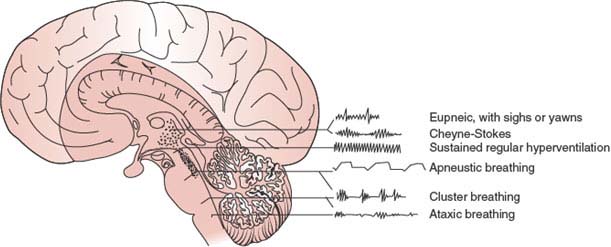
FIG. 23.2. Respiratory patterns characteristic of lesions at different levels of the brain.
CHEYNE–STOKES RESPIRATION
This type of respiration consists of brief periods of hyperpnea alternating regularly with even shorter periods of apnea. After the apneic phase, the amplitude of respiratory movements increases gradually to a peak and then slowly wanes to apnea. During the hyperpneic stage, the patient becomes more alert, the pupils may dilate toward normal from the miosis characteristic of diencephalic dysfunction, and the motor behavior reflects control by higher centers (e.g., decorticate posturing yields to semipurposeful movements). The eyelids may open during the rapid breathing phase and close during the slow breathing phase.
Cheyne–Stokes respiration represents a more severe degree of posthyperventilation apnea in which the respiratory drive becomes more closely dependent on the pCO2. Because the “smoothing effect” provided by forebrain structures has been removed, pCO2 accumulation causes hyperpnea, which in turn induces a drop in pCO2. With this drop, the respiratory stimulus ceases, and a period of apnea ensues.
This respiratory pattern may follow bilateral widespread cortical lesions but is more likely to be associated with bilateral thalamic dysfunction and has also been described with lesions of the descending pathways anywhere from the cerebral hemispheres to the level of the upper pons [106]. Metabolic disturbances, such as uremia, diffuse anoxia, and heart failure, often underlie this breathing disorder. This pattern of respiration may also be seen in some elderly individuals during sleep and in some normal individuals at high altitudes. Cheyne–Stokes respiration in patients with supratentorial mass lesions may indicate incipient transtentorial herniation [118].
HYPERVENTILATION WITH BRAINSTEM INJURY
Patients with lesions of the midbrain and pons often have prolonged and rapid hyperpnea. Because most of these patients are relatively hypoxic despite the excessive ventilatory effort, this type of breathing cannot truly be called neurogenic hyperventilation. In a few cases where pulmonary or metabolic causes of hyperventilation were absent, brainstem tumors were found at autopsy. In these cases, tumoral metabolism may have lowered the pH of the local cerebrospinal fluid, thereby providing a stimulus to the respiratory center of the medulla [117]. Central neurogenic hyperventilation, responsive to morphine and methadone, occurred with an astrocytoma centered in the medial tegmental parapontine reticular formation [56].
APNEUSTIC BREATHING
Apneustic breathing is characterized by a long inspiratory pause, after which the air is retained for several seconds and then released. This abnormality appears with lesions of the lateral tegmentum of the lower half of the pons.
CLUSTER BREATHING
Breathing with a cluster of breaths following each other in an irregular sequence may result from low pontine or high medullary lesions.
ATAXIC BREATHING
This type of breathing has a completely irregular pattern (also called the atrial fibrillation of respiration) in which inspiratory gasps of diverse amplitude and length are intermingled with periods of apnea. This respiratory abnormality, often present in agonal patients, heralds complete respiratory failure and follows damage of the dorsomedial medulla. The most common etiologies for this pattern include cerebellar or pontine hemorrhages, trauma, and posterior fossa tumors. Less often, a paramedian medullary infarct (usually due to severe atherosclerosis of a vertebral artery) may cause this syndrome. The classic breathing pattern described by Biot was ataxic breathing in patients with severe meningitis [118].
“ONDINE CURSE”
Pathways from the cerebral cortex subserving voluntary respiration are separate from those descending from the medulla subserving automatic respiration; thus, selective impairment of automatic or voluntary breathing is possible [156]. Descending pathways that are under voluntary control travel within the dorsal cord in the region of the corticospinal tract, whereas pathways from primary medullary respiratory centers travel in the ventrolateral cord, with anatomic separation of inspiratory and expiratory pathways [156].
Ondine curse refers to the loss of automatic breathing during sleep. This respiratory pattern, obviously absent in comatose patients, is mentioned here because it occurs with lower brainstem dysfunction. Responsible lesions have a similar or somewhat lower location than those that cause ataxic breathing but are smaller or develop more slowly. Both unilateral [5,82] and bilateral [30] medullary tegmental infarcts have produced this syndrome. This disorder has also been recorded with high cervical cord lesions after surgical section of the ventrolateral spinal cord for pain relief [75,170], probably because of reticulospinal tract interruption. Of 12 patients who died with presumed Ondine curse after high cervical percutaneous cordotomy for pain, all had lesions involving the region of the anterolateral funiculus in the C2 segment containing pain fibers activated from the second to fifth thoracic dermatomes [75].
Central hypoventilation may be caused by unilateral caudal brainstem infarction [12]. One patient with nearly complete loss of ventilation involving both automatic and voluntary components had an infarct involving the reticular formation, nucleus tractus solitarius, nucleus ambiguus, and nucleus retroambiguus on the right, which spared the dorsal motor nucleus of the vagus nerve and sensory and corticospinal tracts. A second patient with hypoventilation more selectively involving automatic responses (Ondine curse) had an infarct involving the medullary reticular formation and nucleus ambiguus that spared the nucleus tractus solitarius. These cases suggest that unilateral involvement of the pontomedullary reticular formation and nucleus ambiguus is sufficient for generating loss of automatic respiration, whereas an associated lesion of the nucleus tractus solitarius may lead to more severe respiratory failure involving automatic and voluntary responses [12]. Central hypoventilation or apnea has also been reported with bilateral damage to the high cervical spinal cord or with dorsolateral lesions of the tegmentum of the medulla. A selective paresis of voluntary but not automatic respiration has been described with a discrete infarction of the ventral basis pontis, further suggesting that automatic and voluntary respirations are controlled by anatomically independent pathways [100].
In patients who have lost all respiratory reflexes and are intubated, the self-cycling of the ventilator may erroneously suggest that the patient is triggering it [182].
Temperature Changes
Hyperthermia is not uncommon in coma caused by severe traumatic brain injury [165]. Patients in coma are predisposed to infection, but in a proportion of patients, hyperthermia may be neurogenic, that is, related to an altered temperature regulation system. In many of these instances, they correspond to hypothalamic dysfunction (see Chapter 17). Neurogenic hyperthermia has also been described with pontine tegmental lesions [110].
The Pupils
Pupillary shape, size, symmetry, and response to light provide valuable clues to brainstem and third cranial nerve function. The pupillary light reflex is very resistant to metabolic dysfunction. Abnormalities of this reflex, particularly when unilateral, indicate structural lesions of the midbrain or oculomotor nerve. A few exceptions are noteworthy. Atropinic agents, instilled into the eyes, applied on the skin (e.g., transdermal scopolamine) [24], ingested, or given during cardiopulmonary resuscitation, may cause pharmacologic iridoplegia. In these cases, a solution of 1% pilocarpine applied to the eye will fail to constrict the pupils, whereas in the case of anoxic pupillary dilation, this cholinergic agent, acting directly on the constrictor of the iris, produces miosis. Because many patients in coma have small pupils, anticholinergic agents are sometimes used to facilitate visualization of the optic fundi, thus eliminating a potentially useful diagnostic indicator. In many cases, a better way to obtain pupillary dilation is by pinching the skin of the neck (ciliospinal reflex). Glutethimide (Doriden) induces unequal pupils that are midsized or slightly dilated and poorly responsive to light. Other agents that may cause unreactive pupils include barbiturates (the pupillary light reflex is more often retained), succinylcholine, and, rarely, other anticonvulsants, lidocaine, phenothiazines, methanol, and aminoglycoside antibiotics [25,42]. Agents other than glutethimide or anticholinergic drugs cause pupillary dilation only when taken in massive amounts, enough to eliminate respiratory reflexes or, in the cases of succinylcholine and aminoglycoside antibiotics, generalized neuromuscular junction blockade. Usually, the amount of sedative drug is insufficient to abolish the pupillary light reflex. Hypothermia and acute anoxia may also cause unreactive pupils, which, if persistent beyond several minutes after an anoxic insult, carry a poor prognosis [38].
The areas of the brain and anatomic pathways that mediate the pupillary light reflex are reviewed in Chapter 8, in which the origin and course of sympathetic and parasympathetic influences on the iris muscle are described.
Various structural lesions causing coma may be associated with pupillary abnormalities (Fig. 23.3):
1. Sleep or bilateral diencephalic dysfunction (metabolic coma) is accompanied by small pupils that react well to light (“diencephalic” pupils).
2. Unilateral hypothalamic damage induces miosis and anhidrosis on the side of the body ipsilateral to the lesion.
3. Midbrain lesions causing coma usually produce distinct pupillary abnormalities. Tectal or pretectal lesions affecting the posterior commissure abolish the light reflex, but the pupils, which are midsized or slightly large, may show spontaneous oscillations in size (hippus) and become larger when the neck is pinched (ciliospinal reflex). Tegmental lesions, which involve the third nerve nucleus, may cause irregular constriction of the sphincter of the iris, with a resultant pear-shaped pupil or displacement of the pupil to one side (midbrain corectopia) [151]. The pupils, often unequal, tend to be midsized and lack light or ciliospinal responses. Unilateral or bilateral oval pupils (which may be fixed to light) may occur with severe cerebrovascular lesions that injure the oculomotor or pupillomotor fibers. The oval shape is due to nonuniform paresis or paralysis of the pupil sphincter, with resultant eccentric antagonistic effects of pupil dilators.
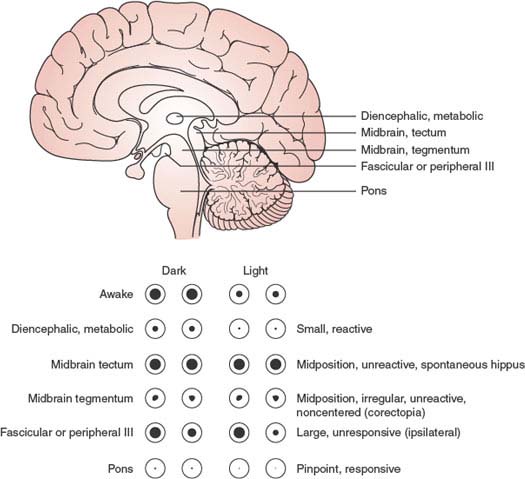
FIG. 23.3. Pupillary responses characteristic of lesions at different levels of the brain.
4. Pontine tegmental lesions cause small pupils due to interruption of descending sympathetic pathways. Pinpoint pupils, when observed with a magnifying glass, may be seen to constrict to light, may occur with pontine hemorrhage, and are due to a combination of sympathetic damage and parasympathetic irritation.
5. Lateral pontine, lateral medullary, and ventrolateral cervical cord lesions produce an ipsilateral Horner syndrome.
6. Oculomotor nerve compression and elongation by herniation of the uncus of the temporal lobe (through the tentorial incisura) affect pupillary function earlier and more noticeably than the extrinsic eye movements subserved by this cranial nerve. Possible explanations for pupillary dilation on the side of a mass lesion include compression of the third cranial nerve by uncal herniation beneath the tentorial edge; compression of the nerve by the posterior cerebral artery or by the hippocampal gyrus; stretching or buckling of the nerve by traction at the superior orbital fissure, posterior clinoid, or clivus; or compression of the midbrain oculomotor complex [137]. The light reflex is sluggish or absent, and, unlike the situation with midbrain involvement, the pupil becomes widely dilated owing to sparing of the sympathetic pathways (Hutchinson pupil).
Ropper studied the pupil opposite the one already enlarged from transtentorial herniation in 13 patients [135]. In most patients, the pupil was initially 2.5 to 4 mm in diameter with a diminished or absent light reaction; this initial phase was followed by a slight reduction in pupil size, then reenlargement to greater than the original pupil size, all with preserved roundness. Subsequent deterioration varied, but a transitional oval pupil shape was infrequent, and oculomotor function was otherwise preserved until both pupils were enlarged and fixed to light. Thus, subsequent neurologic deterioration in a patient with transtentorial herniation can often be appreciated by change in the reactivity and size of the opposite pupil [135]. Other reports have demonstrated a paradoxical initial enlargement of the pupil opposite the side of a mass lesion, especially with acute subdural hematoma [115] or intraparenchymal [22] or subarachnoid [88] hemorrhage.
7. Other oculomotor nerve lesions causing pupillary abnormalities are less likely to impair consciousness, except when associated with a subarachnoid hemorrhage. Posterior communicating artery aneurysms can compress the third nerve and a massive subarachnoid hemorrhage may result in coma. Rarely, with the Guillain–Barré syndrome patients may become completely paralyzed and lose even their pupillary response [8,124]. This complete locked-in state may be mistaken for severe anoxic brain damage in these patients who are obviously prone to anoxic events [80]. However, in a locked-in patient, the electroencephalogram will show normal or slightly slow brain activity.
With acute neurosurgical lesions, fixed pupils are not necessarily a sign of irreversible coma. In a series of 40 patients with fixed pupils, 25% of them made a functional recovery [146]. None of these patients recovered after more than 6 hours with fixed pupils.
Eye Movements
The anatomic pathways subserving eye movements were reviewed in Chapter 8. In the comatose patient, the assessment of eye movements helps to determine the level of structural brainstem damage (Fig. 23.4) or the depth of coma induced by metabolic agents.
In the absence of voluntary eye movements, the assessment of ocular motility in comatose patients relies heavily on reflex eye movements, including the oculocephalic reflex, elicited by the doll’s eye maneuver, and the oculovestibular reflex, elicited by instillation of cold or warm water into the external auditory canal [16,118]. Caloric testing with 50 mL of ice water instilled over 30 seconds into the external auditory canal, after the head is raised 30 degrees and an intact tympanic membrane is documented, provides a stronger stimulus than the oculocephalic reflex. If only the latter reflex is present, either caloric stimulation has been performed inadequately (e.g., hindered by the presence of wax in the external auditory canal) or there is damage to the labyrinth (e.g., by ototoxic antibodies) or the vestibular nuclei in the laterosuperior medulla.
Because of the absence of cortical control of eye movements, the comatose patient lacks voluntary saccades, including the quick phase of nystagmus and tracking eye movements. Instead, if the brainstem is intact, the eyelids are closed, and the eyes, slightly divergent, drift slowly from side to side (roving eye movements). Spontaneous blinking requires an intact pontine reticular formation. Blinking induced by a bright light is probably mediated by the superior colliculus and remains intact despite occipital damage. Absence of blinking only on one side indicates unilateral nuclear, fascicular, or peripheral facial nerve dysfunction. The eyelids may remain tonically retracted due to failure of levator inhibition in some cases of pontine infarction (eyes-open coma) [66].
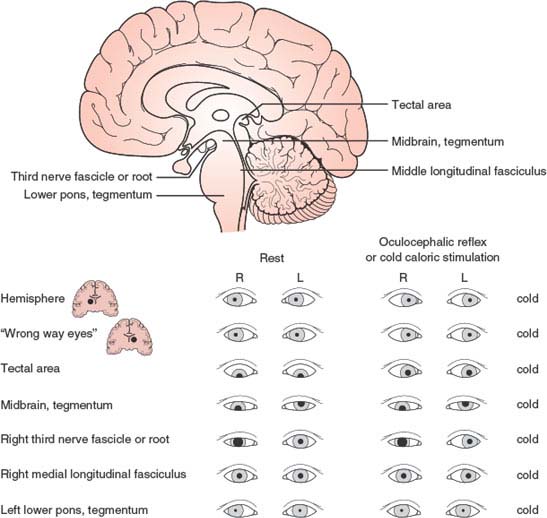
FIG. 23.4. Eye movement abnormalities characteristic of lesions at different levels of the brain. The responses to cold caloric stimulation of the left ear are indicated in the right-hand column. The first two responses at the top show normal extrinsic ocular motility with cold caloric stimulation of the left ear.
The roving eye movements of light coma cannot be voluntarily executed and are therefore incompatible with the diagnosis of feigned unresponsiveness. As coma deepens, roving eye movements disappear first, followed by the oculocephalic reflex; finally, even cold water instilled in the ear fails to induce eye movements. In metabolic coma, the pupils may still react when eye movements cannot be elicited.
Other spontaneous eye movements seen in comatose patients include the following (Table 23.1):
1.Short-cycle periodic alternating gaze (ping-pong gaze), which consists of roving of the eyes from one extreme of horizontal gaze to the other and back, with each oscillating cycle taking 2.5 to 8 seconds [32,54,90,161]. This finding usually indicates bilateral cerebral damage (e.g., bilateral cerebral infarcts) with an intact brainstem, but it has also been described with posterior fossa hemorrhage, basal ganglia infarcts, hydrocephalus, and overdose of the monoamine oxidase inhibitor tranylcypromine [54,77,78,128,153,177]. The disorder may occasionally occur in coma with no structural hemispheric lesion [76,190]. A man in his twenties, with chronic hydrocephalus from infancy and absent vertical eye movements, had ping-pong gaze since childhood only when awake [78]. One case was attributed to bilateral lesions of the cerebral peduncles [76].
TABLE 23.1 Spontaneous Eye Movements in Comatose Patients
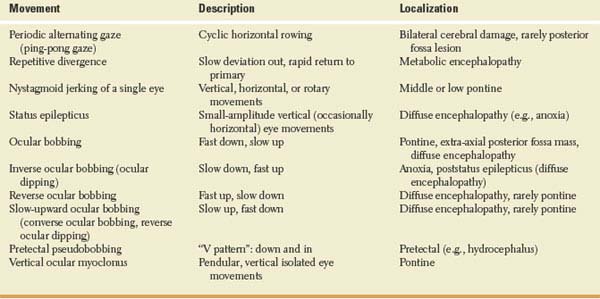
Crevits and Decruyenaere described three patients with ping-pong gaze (due to hepatic encephalopathy, carbon monoxide intoxication, and hypoxia, respectively) and proposed that this term be reserved for those forms of periodic alternating gaze without a silent period [26]. They noted that the only constant clinical implication of ping-pong gaze was integrity, at least in part, of the lower brainstem, with lack of cortical inhibition of the horizontal gaze centers in the brainstem. This disorder of ocular motility had no prognostic value [26].
Ping-pong gaze must be differentiated from periodic alternating gaze deviation, which is an alternating horizontal conjugate gaze deviation lasting 1 to 2 minutes in each direction. Periodic alternating gaze deviation usually occurs in alert patients with structural lesions involving the cerebellum and brainstem, such as the Arnold-Chiari malformation or medulloblastoma, but it has been described in obtunded or comatose patients with hepatic encephalopathy [6].
2.Repetitive divergence is rarely seen in patients with coma from metabolic encephalopathy (e.g., hepatic encephalopathy) [104]. With this disorder, the eyes are midposition or slightly divergent at rest. They then slowly deviate out, become fully deviated for a brief period, and then rapidly return to primary position before repeating the cycle. These motions are synchronous in both eyes.
3.Nystagmoid jerking of a single eye, in a vertical, horizontal, or rotatory fashion, may occur with mid-to-lower pontine damage [118]. Pontine lesions occasionally give rise to disconjugate rotatory and vertical movements of the eyes, in which one eye may rise and intort as the other falls and extorts [118]. This type of movement should not be confused with see-saw nystagmus, which is very seldom seen in comatose patients [64].
4.Electrographic status epilepticus without appendicular motor manifestations, due to anoxia, may result in brisk, small amplitude, mainly vertical (occasionally horizontal) eye movements detectable by passive lid elevation [157].
5.Ocular bobbing refers to intermittent, often conjugate, brisk, bilateral downward movement of the eyes with slow return to midposition [39]. Both mesencephalic and medullary burst neuron centers may play a part in its genesis [138]. Cold calorics may increase the amplitude and frequency of the bobbing or have no effect [27]. Ocular bobbing has been associated with intrinsic pontine lesions (e.g., hemorrhage, tumor, infarction, central pontine myelinolysis) [60,79,93,138,167,190], extra-axial posterior fossa masses (e.g., aneurysm rupture or cerebellar hemorrhage or infarction) [13,48,108], diffuse encephalitis [144], Jakob–Creutzfeldt disease [138], and toxic-metabolic encephalopathies (e.g., acute organophosphate poisoning) [34,49,162]. “Typical” ocular bobbing, which is associated with preserved horizontal eye movements, is thought to be specific but not pathognomonic of acute pontine injury, whereas “atypical” ocular bobbing, which is associated with absent horizontal eye movements, is thought to be less helpful in predicting the site of abnormality [162]. Monocular bobbing (paretic bobbing), which consists of a quick downward movement of one eye and intorsion or no movement in the other eye, may occur if there is a coexistent unilateral fascicular oculomotor nerve palsy [162]. Disconjugate ocular bobbing, with movements involving sometimes one eye and sometimes the other, may also occur without oculomotor nerve palsy [43].
6.Inverse ocular bobbing (ocular dipping or fast-upward ocular bobbing) [44,70,85,93,131,145,159,173] consists of a slow-downward eye movement with fast return to midposition, which may occur in anoxic coma or after prolonged status epilepticus [70,133,159]. It probably reflects diffuse brain dysfunction rather than a single structural lesion because brainstem horizontal gaze reflexes are usually intact. Ocular dipping has also been described associated with deafness in a patient with pinealoblastoma [169]. Inverse/ reverse ocular bobbing consists of inverse ocular bobbing in which the eyes do not stop on rapidly returning to primary position but shoot into upgaze and slowly return to midposition [140,168].
7.Reverse ocular bobbing (fast-upward ocular bobbing) consists of fast-upward eye movement with a slow return to midposition, which may occur in patients with metabolic encephalopathy, viral encephalitis, or pontine hemorrhage [15,44,93]. It has been described with coma, due to combined phenothiazine and benzodiazepine poisoning [81]. Occasionally, ocular bobbing, ocular dipping, and reverse bobbing may occur at different times in the same patient [140].
8.Slow-upward ocular bobbing (converse ocular bobbing or reverse ocular dipping) is characterized by slow-upward eye movements followed by fast return to midposition [44,93]. This eye movement disorder has been described with pontine infarction (the patient had a one-and-a-half syndrome) [44] and with metabolic or viral encephalopathy (i.e., diffuse cerebral dysfunction) [93].
9.Pretectal pseudobobbing has been described with acute hydrocephalus [65] and consists of arrhythmic, repetitive downward and inward (“V-pattern”) eye movements at a rate ranging from 1 per 3 seconds to 2 per second and an amplitude of 1/5 to 1/2 of the full voluntary range. These movements may be mistaken for ocular bobbing, but their V pattern, their faster rate, and their pretectal rather than pontine-associated signs distinguished them from true pontine bobbing. Thus, patients with pretectal pseudobobbing may have abnormal pupillary light reactions, intact horizontal eye movements, open and often retracted eyelids, a blink frequently preceding each eye movement, and a mute or stuporous rather than a comatose state. Pretectal pseudobobbing probably represents a variety of convergence nystagmus, and its presence usually indicates the need for prompt surgical attention (e.g., hydrocephalus decompression) [65]. It is possible that some cases of “ocular bobbing” associated with thalamic hemorrhage or tentorial herniation may actually be cases of pretectal pseudobobbing.
10.Vertical ocular myoclonus consists of pendular, vertical isolated movements of the eyes noted in patients either locked-in or comatose after severe pontine strokes [61]. Their frequency is 2 Hz, and other rhythmic body movements at a similar frequency occur after a 6-week to 9-month delay. These movements are generally associated with palatal myoclonus (palatal tremor), with which they share a common mechanism [61].
ABNORMALITIES OF LATERAL GAZE
Conjugate Gaze. When both eyes remain deviated toward the same side in a comatose patient, the lesion may be in the cerebral hemisphere (most often involving the frontal eye fields) or in the pontine tegmentum. In the case of a hemispheric lesion, unless the patient is having a seizure, the eyes “look toward the lesion” (away from the hemiparetic side) but can be brought to the other side with the oculocephalic maneuver, caloric testing, or both. A seizure originating in the frontal or occipital lobes may cause deviation of the eyes and head away from the lesion, but such deviation is brief and usually accompanied by nystagmoid jerks; as soon as the seizure ceases, the eyes return to “look” toward the lesion. Thalamic and, rarely, basal ganglionic lesions, almost always hemorrhagic, may produce forced deviation of the eyes to the side contralateral to the lesion (wrong-way eyes) [166]. Very rarely, frontal-perisylvian lesions may cause wrong-way eyes [113].
Predominantly unilateral lesions affecting the tegmentum of the lower pons cause a horizontal gaze palsy toward the side of the lesion so that the eyes look toward the hemiparetic side. Neither the oculocephalic maneuver nor caloric testing overcomes a pontine gaze palsy.
Coma due to toxic substances is often accompanied by impaired conjugate eye movements, horizontal as well as vertical [119]. Thiamine deficiency, causing Wernicke encephalopathy, is a treatable cause of ophthalmoparesis and coma. It need not accompany alcoholism [191].
Disconjugate Gaze. Isolated failure of ocular adduction, in the absence of pupillary changes and with normal vertical eye movements (elicited by oculocephalic or oculovestibular reflexes), indicates a lesion of the medial longitudinal fasciculus (MLF) in the upper pons ipsilateral to the eye that fails to adduct. MLF involvement is commonly bilateral in comatose patients. Rarely, metabolic coma (such as that due to barbiturates, amitriptyline [52], or hepatic failure [19]) may induce a transient MLF syndrome that can usually be overcome by vigorous caloric testing.
Latent strabismus may become apparent when the level of alertness is mildly impaired but disappears in deep coma. Because strabismus involves a single muscle, it seldom mimics neurogenic oculoparesis except, perhaps, when abduction is reduced.
ABNORMALITIES OF VERTICAL GAZE
In patients in light coma, upward gaze can be tested by holding the eyelids open and gently touching the cornea with a wisp of cotton or a similar object. With this stimulus, the eyeballs tend to roll upward (Bell’s phenomenon). Unless the patient is intubated or has a neck injury, the doll’s head maneuver can be used to elicit the vertical component of the oculocephalic reflex. Irrigation of both ears with cold water induces downward deviation of the eyes; warm water induces upward deviation.
Disconjugate vertical gaze in the resting position (skew deviation) may be seen with lesions at different areas of the brainstem, with increased intracranial pressure, or with hepatic coma (see Chapter 8). Persistent deviation of the eyes below the horizontal meridian signifies brainstem dysfunction, which is often due to a structural lesion that affects the tectum of the midbrain but is occasionally caused by metabolic encephalopathy (e.g., hepatic coma [69]). It is also present frequently after anoxic brain damage caused by cardiac arrest, typically a few days after the event [57]. Tonic downward deviation of the eyes, often accompanied by convergence, may occur with thalamic hemorrhage, probably due to pressure on the dorsal mesencephalon. Forced downward deviation of the eyes has also been reported in patients feigning coma [139]. Forced downward deviation of the eyes during caloric testing often occurs in coma induced by sedative drugs [155]. Tonic upgaze has been reported shortly after severe anoxic encephalopathy [57,67] and with phenothiazine intoxication. Paresis of upward gaze is usually present with bilateral midbrain tectal damage. Downward gaze is preferentially affected by bilateral lesions of the superomedial perirubral region in the ventral portion of the origin of the Sylvian aqueduct from the third ventricle. In many of these patients, the lesion extends into the medial thalamic nuclei [112]. Large midbrain tegmental lesions abolish vertical gaze. At rest, the eyes remain in midposition or may be disconjugately deviated in the vertical plane.
Bilateral ptosis newly developed in patients with evolving massive hemispheric infarction seems to suggest compression of the midbrain [11].
Corneal Reflex
The corneal reflex has a higher threshold in comatose patients. Nonetheless, it must be elicited by a gentle and aseptic stimulus to avoid the risk of an infected corneal ulceration in patients with decreased corneal sensitivity (as with cranial nerve V lesions, ipsilateral lateral pontomedullary lesions, or contralateral parietal lesions) [107], or impaired eye closure (as with cranial nerve VII lesions and low pontine lesions). In the latter cases, the stimulus may induce deviation of the jaw to the opposite side (corneopterygoid reflex), and, given an intact upper pons and midbrain, the eyes may roll upward (Bell’s phenomenon).
Motor Activity of the Body and Limbs
When examining a patient in coma, observation of the movements and of the tone and reflexes of the limbs supplies information that has a less clear-cut localizing value than similar findings in alert patients. Rarely is a metabolic coma (notably hypoglycemic) accompanied by hemiparesis; however, other motor patterns, widely known as decorticate (flexor posturing) and decerebrate (extensor posturing) rigidity, are often produced by metabolic disorders [46] and do not have the structural implications that their names, coined during experimental work, would suggest. Of course, structural damage to the origin or course of the motor pathways may give rise to such patterns, which, in these cases, are often asymmetric. However, because metabolic coma is more frequent than structural coma, metabolic coma is often responsible for the motor patterns discussed below (Fig. 23.5).
In light coma, the general motor responses may oscillate between lying quietly in bed and wildly thrashing about. The latter situation occurs when a painful stimulus (such as that caused by a subarachnoid hemorrhage or a full bladder) rouses the patient, whose diminished attention prevents any coherent sequence of movements. Such patients, however, try to avoid painful stimuli by appropriately withdrawing a limb or using it to brush off the offending agent. Gently sliding a cotton-tipped stick along the patient’s forehead often proves enough of a stimulus to obtain such a response. Asymmetric responses betray a deficit of the motor or sensory pathways, or both.
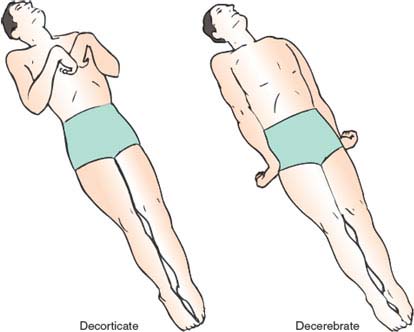
FIG. 23.5. Decorticate and decerebrate posturing of the limbs in comatose patients.









