Chapter 19 The tardive syndromes
Phenomenology, concepts on pathophysiology and treatment, and other neuroleptic-induced syndromes
Overview
Fundamentals and definitions
A variety of neurologic adverse effects are seen with drugs that block dopamine D2 receptors. Because these complications are mainly movement disorders and likely relate to the D2 receptors in the striatum and limbic system, they are usually called extrapyramidal reactions. These are listed in Table 19.1 and are covered in the clinical sections that follow.
Table 19.1 Neurological adverse effects of dopamine receptor antagonists
Adapted from Fahn S: The tardive dyskinesias. In Matthews WB, Glaser GH (eds): Recent Advances in Clinical Neurology, vol. 4. Edinburgh: Churchill Livingstone, 1984, pp. 229–260.
The term tardive syndromes refers to a group of disorders that fit all of the following essential criteria: (1) Phenomenologically, the clinical features are that of a movement disorder – i.e., abnormal involuntary movements or a sensation of restlessness that often causes “unvoluntary” movements; (2) the disorder is caused by the patient’s having been exposed to at least one DRBA within 6 months of the onset of symptoms (in exceptional cases, exposure could be up to 12 months); and (3) the disorder persists for at least 1 month after stopping the offending drug (Fahn, 1984a; Stacy and Jankovic, 1991). The question arises as to what to call persistent dyskinesias that are induced by drugs other than DRBAs. For example, Miller and Jankovic (1992) described such a patient who had been exposed to flecainide, a drug not known to be a DRBA. Another drug, buspirone, an azospirone compound, is an anxiolytic that is not known to have any dopamine receptor-blocking activity. Yet there is a report of two patients who had persistent movement disorders after prolonged treatment with this drug (LeWitt et al., 1993). One patient had cervical-cranial dystonia, and the other had an exacerbation of preexisting spasmodic torticollis and TD. It is possible, however, that future laboratory investigation will reveal flecainide and buspirone or one of their metabolites actually to be a DRBA.
Although there are several phenomenologically distinct types of tardive disorders, the collective group is referred to as tardive dyskinesia for historical reasons. Unfortunately, the term tardive dyskinesia is often used also to refer to a phenomenologically specific type of tardive syndrome, so the literature is often confusing; this note of caution is particularly important in trying to understand whether the author of a paper is referring to the tardive syndromes collectively or to a specific tardive syndrome. Since there could be different pathophysiologic mechanisms and treatments for the different forms of tardive syndromes, it is best that they be divided phenomenologically (Table 19.2). In this review, the tardive syndromes as a whole are referred to as tardive dyskinesia, and the specific type that was historically and initially labeled as tardive dyskinesia is referred to as classic tardive dyskinesia. Other names have been used for classic tardive dyskinesia (see Table 19.2). The phenomenologically essential component of classic tardive dyskinesia is the presence of repetitive, almost rhythmic, movements. These are almost always present in the mouth region, and therefore are also called O-B-L dyskinesias.
Table 19.2 Terminology of the tardive syndromes
| Descriptions | Equivalent common names |
|---|---|
| Tardive syndromes as a group | Tardive syndrome |
| Tardive dyskinesia | |
| Repetitive, rhythmic movements, usually in the oral-buccal-lingual region | Classic tardive dyskinesia (TD) |
| Oral-buccal-lingual (O-B-L) dyskinesias | |
| Tardive stereotypy | |
| Rhythmic chorea | |
| Dystonic movements and postures | Tardive dystonia |
| Restlessness and the movements that occur as a result | Tardive akathisia |
| Myoclonus | Tardive myoclonus |
| Tremor | Tardive tremor |
| Tics | Tardive tics |
| Tardive tourettism | |
| Chorea | Withdrawal emergent syndrome |
| Tardive chorea | |
| Oculogyria | Tardive oculogyric crisis |
| Parkinsonism | Tardive parkinsonism (if it exists) |
Tardive dyskinesia was first described in patients who were treated for schizophrenic psychosis with antipsychotics (Schonecker, 1957; Sigwald et al., 1959). These abnormal movements appeared late in the course of treatment, in contrast to acute dystonic reactions and drug-induced parkinsonism, which had previously been recognized as complications from antipsychotics, hence the term tardive. The offending antipsychotics are now known to block the D2 dopamine receptor; i.e., these drugs are DRBAs. Since the first descriptions, TD has also been noted in patients without psychiatric disorders who had other indications for using dopamine receptor antagonists, such as those with gastrointestinal complaints (Casey, 1983), with Gilles de la Tourette syndrome (Riddle et al., 1987), or with dystonia (Greene and Fahn, 1988).
Dopamine receptor antagonists produce many undesirable side effects, most of which occur relatively early in the course of treatment and are reversible on discontinuation of the medication. However, disfiguring and disabling abnormal involuntary movements were also noted to often occur late in the course of treatment and these were often noted to persist, even after discontinuation of the medication. Hence, the term “tardive” was coined, referring to the late and insidious onset (Faurbye et al., 1964). Initially the term tardive dyskinesia was equated with stereotypic repetitive movements of oral, buccal, and lingual distribution (Schonecker, 1957), but subsequently other types of movements have been recognized (Burke et al., 1982; Fahn, 1984a). As such, the concept of tardive dyskinesia has evolved and has been modified considerably since the initial recognition of the syndrome.
The prevalences of drug-induced parkinsonism and the various tardive syndromes have been compared by van Harten and colleagues (1996b) on the island of Curaçao, which has only one psychiatric facility. In 194 inpatients, the prevalence for classic tardive dyskinesia (TD) was 39.7%, that for parkinsonism was 36.1%, that for tardive dystonia was 13.4%, and that for akathisia was 9.3%. Combinations of two or more of these phenomenologies occurred in 30% of patients (van Harten et al., 1997). van Harten and colleagues (2006) continued to follow their patients over 9 years (mean duration of 18 years exposure to first-generation antipsychotic drugs); they found the annual incidence rate for classic TD to be 10.2% and for tardive dystonia to be 0.7%. Severity was associated with age and akathisia but not with drug-induced parkinsonism.
Dopamine receptors and their antagonists
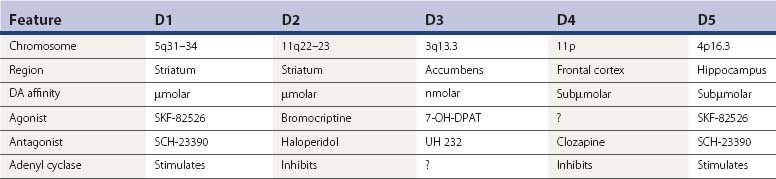
Since TD is an iatrogenic disorder and the most constant feature of the syndrome is the pharmacologic class of the responsible etiologic agent, it is important to understand the nature of the drugs that produce TD (Table 19.3). Although models of abnormal basal ganglia circuitry have been proposed to explain the mechanism of TD (Marchand and Dilda, 2006), the pathophysiology is still poorly understood. Dopamine receptors are classified into five subtypes, based on the genetics of the receptors; they are labeled D1, D2, D3, D4, and D5 (Kebabian and Calne, 1979; Sokoloff et al., 1990). Table 19.4 characterizes the five dopamine receptors. It is the dopamine D2 receptor-blocking action of drugs that has been linked to the tardive syndromes and other neuroleptic drug-induced movement disorders (described later and listed in Table 19.5).
Table 19.3 Drugs that can produce tardive syndromes
| Class of drug | Examples of drugs in each class |
|---|---|
| 1. Phenothiazines | |
| a. Aliphatic | Chlorpromazine (Thorazine) Triflupromazine (Vesprin) |
| b. Piperidine | Thioridazine (Mellaril) Mesoridazine (Serentil) |
| c. Piperazine | Trifluoperazine (Stelazine) Prochlorperazine (Compazine) Perphenazine (Trilafon) Fluphenazine (Prolixin) Perazine |
| 2. Thioxanthenes | |
| a. Aliphatic | Chlorprothixene (Tarctan) |
| b. Piperazine | Thiothixene (Navane) |
| 3. Butyrophenones | Haloperidol (Haldol) Droperidol (Inapsine) |
| 4. Diphenylbutylpiperidine | Pimozide (Orap) |
| 5. Dibenzazepine | Loxapine (Loxitane) Asenapine (Saphris) |
| 6. Dibenzodiazepine | Clozapine (Clozaril) Quetiapine (Seroquel) |
| 7. Thienobenzodiazepine | Olanzapine (Zyprexa) |
| 8. Pyrimidinone | Risperidone (Risperidal) Paliperidone ER (9-hydroxyrisperidone) |
| 9. Benzisothiazole | Ziprasidone (Geodon) |
| 10. Benzisoxazole | Iloperidone (Zomaril) |
| 11. Substituted benzamides | Metoclopramide (Reglan) Tiapride (Tiapridex) Sulpiride (Meresa) Clebopride Remoxipride Veralipride (Agreal, Agradil) Amisulpride (Solian) Levosulpiride |
| 12. Indolones | Molindone (Moban) |
| 13. Quinolinone | Aripiprazole (Abilify) |
| 14. Tricyclic | Amoxapine (Asendin) |
| 15. Calcium channel blockers | Flunarizine (Sibelium) Cinnarizine (Stugeron) |
| 16. N-acetyl-4-methoxytryptamine | Melatonin |
| 17. Interferon-alpha | Pegylated interferon alpha 2b (IFN-α) (Pegylated = polyethylene glycol (PEG) attached to proteins) |
Recently introduced drugs in Table 19.3, e.g., iloperidone (Jain, 2000), ziprasidone (Hirsch et al., 2002), amisulpride (Curran and Perry, 2002), aripiprazole (Tamminga and Carlsson, 2002), paliperidone ER (Chwieduk and Keating, 2010), levosulpiride (Shin et al., 2009), and asenapine (Kane et al., 2010), need to be in clinical use for several years before their full potential in causing tardive dyskinesia syndromes can be known. The earlier ones in the above sentence have already been found to induce the acute and delayed movement disorders described in this chapter. In Table 10.3, the calcium channel blockers deserve comment. Cinnarizine (1-diphenylmethyl-4-(3-phenyl-2-propenyl) piperazine) and its difluorinated derivative flunarizine can induce parkinsonism (Teive et al., 2004). They are antagonists at the D2 receptors (Belforte et al., 2001), and they also inhibit the MgATP-dependent generation of a transmembrane proton electrochemical gradient and dopamine vesicular uptake (Terland and Flatmark, 1999). Whether either of these latter mechanisms, rather than a proposed DRBA action, is responsible for the neuroleptic activity is uncertain. In regard to melatonin, there is a case report of withdrawal-emergent O-B-L dyskinesias associated with akathisia that occurred with sudden discontinuation of chronic melatonin use (Giladi and Shabtai, 1999). Such withdrawal syndromes are typical of drugs that block dopamine receptors. On resumption of melatonin, the patient’s O-B-L dyskinesia and akathisia cleared. Sudden cessation of the drug again brought on the symptoms; slow taper over 2 months was effective without incident. This case suggests that either the melatonin product the patient was taking was impure and was contaminated with a DRBA or melatonin itself has dopamine receptor antagonist properties. In support of the latter is the result of a blinded crossover study showing some suppression of the tardive dyskinesia with melatonin (Shamir et al., 2001). Pegylated interferon alpha-2b (IFN-α), used in the treatment of hepatitis C virus infection, has been reported to cause parkinsonism, akathisia, and acute dystonic reaction (Quarantini et al., 2007). IFN-α has been shown to decrease dopaminergic activity in mice (Shuto et al., 1997).
Some drugs that block dopamine D2 receptors are promoted for medical problems other than psychosis, but these drugs can cause drug-induced parkinsonism, acute dystonic reactions, tardive syndromes, and NMS, just like the drugs that are promoted for the treatment of psychosis. Metoclopramide (Ganzini et al., 1993) and clebopride (Sempere et al., 1994) are used mainly for dyspepsia and as antiemetic agents. Amoxapine has a tricyclic structure and is marketed as an antidepressant drug, but a metabolite has dopamine receptor-blocking activity and has been implicated in producing TD (Kang et al., 1986; Sa et al., 2001). Veralipride is a substituted benzamide that is used for the treatment of menopausal hot flushes (Masmoudi et al., 1995). Pimozide is marketed for the treatment of Tourette syndrome. Some commercial preparations contain dopamine receptor antagonists in combination with other drugs, and this can lead to inadvertent use of these drugs. A popular combination is that of perphenazine and amitriptyline, marketed as Triavil and Etrafon. Risperidone is commercially promoted with the suggestion that it might have less risk of drug-induced complications, but this appears not to be the case. Parkinsonism and TD have been noted in association with the calcium channel antagonists flunarizine and cinnarizine (Micheli et al., 1987). Both of these medications have mild dopamine receptor antagonist activity, which is thought be the mechanism for their complications (Micheli et al., 1989). Recognition of these drugs is essential not only in making the diagnosis of TD but also in preventing the occurrence of TD by being able to avoid using them.
The D1 and D2 receptors are found mainly in the striatum and nucleus accumbens, as well as in the substantia nigra, amygdala, cingulate cortex, and entorhinal area. The anterior lobe of the pituitary gland has only D2 receptors, and the thalamus and cerebral cortex outside of the cingulate and entorhinal area contain D1 receptors only (De Keyser et al., 1988). D2 receptor affinities of dopamine receptor antagonists correlate closely with antipsychotic and antiemetic properties of the drugs (Creese et al., 1976). Dopamine receptor antagonists are often referred to as neuroleptics or antipsychotics; the former term indicates the effect of drugs in producing parkinsonism, and the latter indicates the effect of controlling psychosis.
“Atypical” antipsychotics
The label “atypical” refers to a lower propensity of the antipsychotic agent to induce parkinsonism or a variety of other movement disorders described later (and listed in Table 19.5), that is these agents have a lower propensity to be neuroleptics. A number of epidemiologic studies have found that the atypicals are less likely to induce tardive dyskinesia and other movement disorder problems (Tarsy and Baldessarini, 2006). Dolder and Jeste (2003) found that the atypicals reduced the incidence of tardive dyskinesia by half.
Dopamine and serotonin (5-HT) receptor antagonism
Although drugs that are labeled as atypical antipsychotic agents block dopamine D2 receptors, they also block serotonin 5-HT2A receptors, and some investigators attribute their antipsychotic effect to this mechanism and recommend research and drug development on newer agents that block other 5-HT receptors (Meltzer, 1999).
How tightly drugs to bind to the D2 receptor (their occupancy rate) is currently considered to be important in the drug having neuroleptic potential in direct proportion. Seeman and Tallerico (1998) found that the atypical antipsychotic drugs bind more loosely than classic antipsychotic drugs (neuroleptics) to the D2 receptors. Kapur and colleagues (1999) measured D2 and 5-HT2 receptor occupancies by positron emission tomography (PET) scan for patients receiving clozapine, risperidone, or olanzapine. Clozapine showed a much lower D2 occupancy (16–68%) than risperidone (63–89%) and olanzapine (43–89%); all three showed greater 5-HT2 occupancy than D2 occupancy at all doses, although the difference was greatest for clozapine. In their PET study, Moresco and colleagues (2004) found that striatal D2 receptor occupancy was significantly higher with olanzapine than with clozapine. All these studies support the relationship between receptor occupancy and the clinical observations that clozapine and quetiapine are the most “atypical,” with the least propensity to cause parkinsonism, tardive syndromes, or other neuroleptic drug reactions.
Despite interest in the 5-HT2A receptor, it is the D2 receptor that is associated with the antipsychotic effect (Seeman, 2010). Antipsychotic clinical doses correlate with their affinities for this receptor. The receptor has high- and low-affinity states. Clozapine and quetiapine do not elicit parkinsonism and rarely result in tardive dyskinesia because they are released from D2 within 12–24 hours. Traditional antipsychotics remain attached to D2 receptors for days, preventing relapse, but allowing accumulation that can lead to tardive dyskinesia. Glutamate agonists that treat schizophrenia have affinity for the dopamine D2(High) receptor and the D3 receptor (Seeman and Guan, 2009).
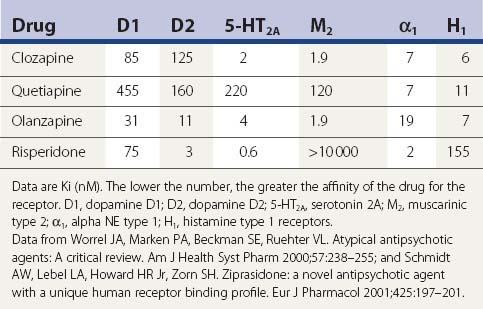
A comparison of the receptor-binding profile of quetiapine, clozapine, olanzapine, and risperidone is presented in Table 19.5. Both quetiapine and clozapine have poor affinity for the dopamine D2 receptor, which probably accounts for the low incidence of inducing parkinsonism and tardive syndromes. Risperidone has the highest affinity for the D2 receptor, resembling haloperidol (see Table 19.6), and is therefore more of a “typical” than an “atypical” antipsychotic.
Table 19.6 A more complete listing of affinities for human receptors and rat transporters by antipsychotic drugs
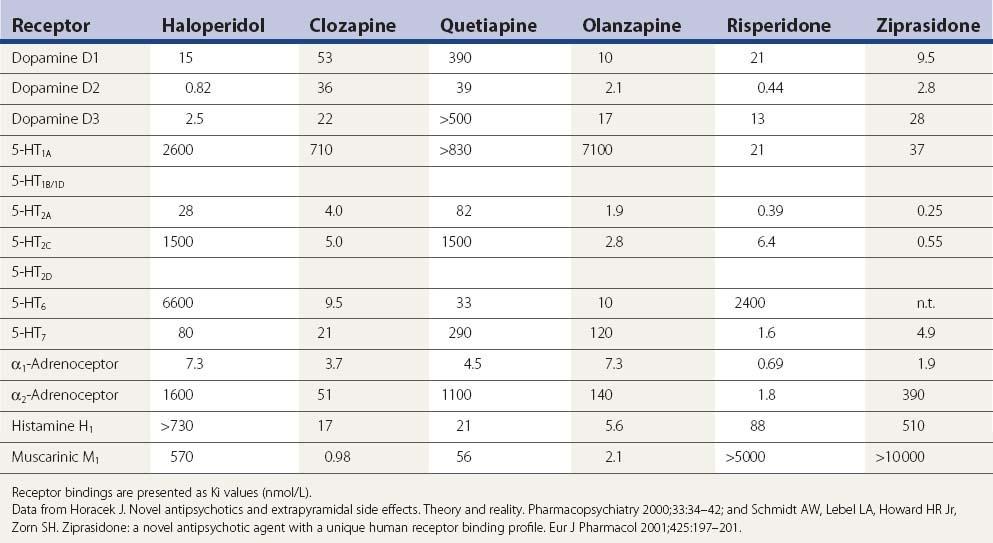
In one PET study striatal D1 and D2 receptor occupancies were evaluated (Tauscher et al., 2004). D1 occupancy ranged from 55% with clozapine to 12% with quetiapine (rank order: clozapine > olanzapine > risperidone > quetiapine). The striatal D2 occupancy ranged from 81% with risperidone to 30% with quetiapine (rank order: risperidone > olanzapine > clozapine > quetiapine). The ratio of striatal D1/D2 occupancy was significantly higher for clozapine (0.88) relative to olanzapine (0.54), quetiapine (0.41), or risperidone (0.31). In an [123I]epidepride single photon emission computed tomography (SPECT) study involving clozapine-, olanzapine-, and haloperidol-treated schizophrenia patients, as well as drug-naive patients and healthy controls, midbrain D(2/3) receptor occupancy was studied. Of those on medication, occupancy was least for those on clozapine (5%), next for those on olanzapine (28%), and greatest for those on haloperidol (40%); no significant differences were observed in the temporal poles (Tuppurainen et al., 2009). All imaging studies are compatible with the clinical results in that clozapine and quetiapine, with the least neuroleptic features, have the lowest D2 receptor occupancy rate.
Clozapine
Clozapine was the first agent to be labeled as an atypical antipsychotic and deservedly so, although rare case reports do exist of drug-induced parkinsonism (Kurz et al., 1995), but it has been reported not to induce rigidity, and it rarely induces parkinsonian tremor (Gerlach and Peacock, 1994). It has also caused rare cases of acute akathisia (Friedman, 1993; Safferman et al., 1993), acute dystonic reaction (Kastrup et al., 1994; Thomas et al., 1994), tardive syndromes (Dave, 1994), tardive dystonia (Bruneau and Stip, 1998; Molho and Factor, 1999a, van Harten et al., 2008b), tardive akathisia (Kyriakos et al., 2005), tardive oculogyria (Uzun and Doruk, 2007), and NMS (Sachdev et al., 1995; Benazzi, 1999; Lara et al., 1999; Gambassi et al., 2006). There are also parkinsonian features that are seen with clozapine. In a prospective study, seven out of 25 patients on clozapine developed TD (Bunker et al., 1996). In a retrospective study, comparison of clozapine and typical antipsychotics showed no lower prevalence of tardive dyskinesia in the clozapine group (Modestin et al., 2000). But it is not clear that the patients in the clozapine group had not been previously exposed to typical antipsychotics and had not developed TD while receiving them. What is clear from this study is that the conversion to using clozapine has not markedly reduced the prevalence of extrapyramidal syndromes; this finding supports the realization that clozapine does not effectively treat TD once it has developed. How effective its use would be in preventing TD in the first place still needs to be verified. For patients with PD, the low propensity to augment existing parkinsonism makes clozapine very useful in treating patients who have dopaminergic drug-induced psychosis (Factor and Friedman, 1997; Friedman et al., 1999).
A SPECT study measuring dopamine D2 receptor binding reveals lower binding with clozapine than with typical neuroleptics (Broich et al., 1998). In animal studies, rats that have been pretreated with haloperidol for 4 weeks develop vacuous chewing movements (VCMs) after treatment with a dopaminergic, whereas rats that have been pretreated with clozapine do not (Ikeda et al., 1999).
A problem with clozapine is that weekly blood counts are required because there is a 1% to 2% incidence of leukopenia, which is reversible if the drug is withdrawn within 1–2 weeks. Granulocyte colony-stimulating factor can be an effective means to treat the agranulocytosis (Sperner-Unterweger et al., 1998). Other common adverse effects are drowsiness, drooling, weight gain, and seizures. An unusual adverse effect with clozapine was a case of myokymia (David and Sharif, 1998). Myocarditis associated with clozapine has been reported (Hill and Harrison-Woolrych, 2008).
Quetiapine
After clozapine, quetiapine is the most favorable in being least likely to induce parkinsonism, NMS (Stanley and Hunter, 2000), or tardive syndromes. In a prospective, head-to-head comparison with risperidone treatment in schizophrenia, quetiapine had far fewer extrapyramidal adverse effects, but more orthostasis (Perez et al., 2008). D2 receptor antagonism is relatively selective for limbic than striatal receptors for clozapine and sertindole, followed by quetiapine, ziprasidone, olanzapine, and remoxipride, whereas risperidone in many respects has a profile that resembles that of haloperidol (Arnt and Skarsfeldt, 1998). In primates, quetiapine and clozapine were equally less likely to induce oral dyskinesias compared to standard antipsychotics (Peacock and Gerlach, 1999). Like clozapine, quetiapine easily induces drowsiness, so when it is used to treat dopa-induced psychosis, it should be taken at bedtime. The major advantage over clozapine is that it does not require blood tests because it does not induce agranulocytosis. However, there may be a rare risk that quetiapine can cause agranulocytosis (Ruhe et al., 2001). Since its introduction, there have been reports of acute dystonic reactions (Jonnalagada and Norton, 2000; Desarker and Sinha, 2006), acute akathisia (Prueter et al., 2003), neuroleptic malignant syndrome (El-Gaaly et al., 2009; Gortney et al., 2009), and also TD (Ghaemi and Ko, 2001). In 367 patients treated for 12 months with quetiapine (mean dose 720 mg/day), parkinsonism was seen in 10%, akathisia in 3%, dystonia in 1.4%, and “hyperkinesia” in 1.7% (Perez et al., 2008).
In a SPECT study using the D2/D3 ligand [I-123]-epidepride the percent occupancy of receptors in the limbic system (temporal lobe) and striatal receptors while patients were receiving quetiapine was 60% and 32%, respectively, which is similar to clozapine (Stephenson et al., 2000). In another SPECT study, quetiapine was shown to occupy 5-HT2A receptors in the frontal and temporal cortex (Jones et al., 2001). A PET study using [18F]fallypride showed similar results (Vernaleken et al., 2010).
A patient with oculogyric crisis, who failed to improve after withdrawal of antipsychotic medication, was successfully treated with quetiapine (Gourzis et al., 2007). Drug-induced parkinsonism and acute akathisia due to other neuroleptics can be reduced by switching patients to quetiapine (Cortese et al., 2008). Drowsiness and weight gain are common; postural hypotension is not infrequent. A rare complication of quetiapine is hepatotoxicity (Shpaner et al., 2008).
Olanzapine
In contrast to clozapine and quetiapine, olanzapine more readily increases parkinsonian symptoms in patients with PD (Jimenez-Jimenez et al., 1998; Molho and Factor, 1999b; Granger and Hanger, 1999), but does so less readily than risperidone and conventional antipsychotics. Drug-induced parkinsonism, including the rabbit syndrome, is seen with olanzapine (Durst et al., 2000), but less than with haloperidol (Peuskens et al., 2009). It can induce acute akathisia (Kurzthaler et al., 1997; Jauss et al., 1998) and TD but less so than haloperidol (Tollefson et al., 1997; Wood 1998). It can also induce NMS (Filice et al., 1998; Moltz and Coeytaux, 1998; Burkhard et al., 1999; Levenson, 1999; Margolese and Chouinard, 1999; Sierra-Biddle et al., 2000; Abu-Kishk et al., 2004; Zaragoza Fernandez et al., 2006), tardive dyskinesia (Herran and Vazquez-Barquero, 1999), and tardive dystonia (Dunayevich and Strakowski, 1999). Acute dystonic reactions also occur (Beasley et al., 1997; Landry and Cournoyer, 1998; Vena et al., 2006).
A direct comparison with chlorpromazine showed similar parkinsonism and acute akathisia for the two drugs (Conley et al., 1998). A double-blind comparison with haloperidol by Beasley and colleagues (1999) showed a much lower risk of developing tardive dyskinesia with olanzapine. After 1 year of exposure, 0.52% of patients developed TD with olanzapine and 7.45% developed TD with haloperidol. An open-label comparison with conventional antipsychotics after a 9-month follow-up after discharge from the hospital favored olanzapine, with TD being present in 2.3% for olanzapine (2/87), and 16.7% (12/72) for the conventional treatment (Mari et al., 2004). There are case reports of agranulocytosis induced by olanzapine (Meissner et al., 1999; Naumann et al., 1999; Tolosa-Vilella et al. 2002), including in a patient who had previously had agranulocytosis with clozapine (Benedetti et al., 1999). This has been attributed to some similar structural and pharmacologic properties of clozapine. A case of restless legs syndrome with periodic movements in sleep has been attributed to olanzapine (Kraus et al., 1999).
Olanzapine has relative regional mesolimbic dopaminergic selectivity and a broad-based binding affinity for serotonin (all 5-HT2 receptor subtypes and the 5-HT6 receptor), dopamine (D2, D3, and D4 receptors), muscarinic, and α1-adrenergic receptors (Bymaster et al., 1999). A PET study in schizophrenic patients being treated with olanzapine revealed that this drug is a potent 5-HT2 blocker, but also a blocker of D2 dopamine receptors similar to risperidone and less so than clozapine (Kapur et al., 1998). Patients on olanzapine were also studied with [123I]iodobenzamide (IBZM) SPECT; the D2 receptor was occupied 60% and 83% of the time at doses of 5 mg/day and 10 mg/day, respectively (Raedler et al., 1999). Such high rates of occupancy probably account for olanzapine’s tendency to worsen parkinsonism and induce DRBA complications, because, as was noted above, D2 receptor occupancy rates are directly related to neuroleptic potential.
Risperidone
With the success of clozapine, there is a commercial advantage for pharmaceutical companies to tout other antipsychotics as “atypical.” Such has been claimed for risperidone, but this drug can readily induce parkinsonism including the rabbit syndrome (Levin and Heresco-Levy, 1999), tardive syndromes (Haberfellner 1997; Silberbauer 1998; Hong et al., 1999; Ananth et al., 2000), and NMS (Newman et al., 1997; Norris et al., 2006). The prevalence of parkinsonism from risperidone is usually considered to be less than that with conventional neuroleptics, but it was observed in 42% compared to 29% in those on haloperidol (Knable et al., 1997). Tardive dyskinesia and tardive dystonia occurred in a patient who was exposed only to risperidone (Bassitt and Garcia, 2000). The annual incidence of TD in patients taking risperidone has been estimated to be 0.3%, compared to an annual incidence in patients taking conventional neuroleptics of 5–10% (Gutierrez-Esteinou and Grebb, 1997). However, in an open-label prospective study of 255 institutionalized patients with dementia who were treated with risperidone, the 1-year cumulative incidence of TD was 2.6% (Jeste et al., 2000).
Like conventional neuroleptics, risperidone induces acute dystonic reactions in marmosets, in contrast to clozapine, which does not (Fukuoka et al., 1997). In a report of an open-label comparison with haloperidol, Rosebush and Mazurek (1999) found the two drugs to have a similar side effect profile. In a prospective follow-up of first-episode schizophrenic patients treated with risperidone, movement disorders developed in more than one-third of these patients, who had previously never been exposed to antipsychotic drugs (Lang et al., 2001). When risperidone is compared with low-potency antipsychotics, such as thioridazine, no difference was discerned in the rates of developing movement disorders (Schillevoort et al., 2001).
In a SPECT study dopamine receptor binding with IBZM showed risperidone to have effects between those of haloperidol and clozapine, with a dose–response curve for risperidone showing greater similarity to that of haloperidol (Dresel et al., 1998). A PET study showed dopamine D2 receptors occupancies of about 70% and 60% in the striatum and extrastriatum, respectively (Ito et al., 2009). Clearly, risperidone is not an “atypical” antipsychotic agent. Risperidone’s occupancy of the 5-HT2 receptors is about 90%, and its occupancy of the D2 receptors is between 50% and 80%, but the latter correlates with the extrapyramidal side effects (Yamada et al., 2002).
Ziprasidone
This benzisothiazole was approved by the Food and Drug Administration in 2001. There are already case reports of acute dystonic reactions (Weinstein et al., 2006; Yumru et al., 2006, Rosenfield et al., 2007), NMS, rhabdomyolysis, and pancreatitis (Murty et al., 2002; Yang and McNeely, 2002; Gray 2004). It has been reported to cause tardive dyskinesia (Mendhekar, 2005) and tardive dystonia (Papapetropoulos et al., 2005; Tsai et al., 2008). Although it is a potent 5-HT2A antagonist (like risperidone, olanzapine, and clozapine), it is also a D2 antagonist in humans as detected by PET scan (Bench et al., 1993, 1996). But in-vitro studies reveal much lower affinity for the D2 receptor than for the 5-HT2A receptor (Seeger et al., 1995), and ziprasidone also binds less tightly to the D2 receptor than dopamine (Seeman, 2002). Ziprasidone has two other unique features compared to other antipsychotic agents: (1) it is a potent 5-HT1A agonist and thus inhibits dorsal raphe serotonergic cell firing (Sprouse et al., 1999) and increases cortical dopamine release (Rollema et al., 2000), and (2) it inhibits neuronal uptake of 5-HT and norepinephrine in a manner comparable to the antidepressant imipramine (Schmidt et al., 2001). What these unique actions might contribute to antipsychotic activity or to propensity for or against extrapyramidal reactions is unclear. Ziprasidone has been reported to induce a case of oculogyric crisis in an adult (Viana Bde et al., 2009).
Aripiprazole
This quinolinone has a novel mechanism of action. Like a number of other “atypical” antipsychotics, it is an antagonist at the 5-HT2A receptors. It is also a 5-HT1A partial agonist (Jordan et al., 2002). What is novel is that it is a partial agonist at the dopamine D2 receptor. It has a higher affinity for the presynaptic autoreceptor than for the postsynaptic receptor. Hence, it reduces dopamine synthesis and release through an agonist action at the dopamine autoreceptor (Tamminga and Carlsson, 2002). Because of its novel action as a partial D2 agonist, it was anticipated that it might cause fewer extrapyramidal adverse effects, and clinical trials reported favorable results (Kane et al., 2002). However, D2 ligand binding PET revealed a dose-related high occupancy state of 71.6% at 2 mg/day to 96.8% at 40 mg/day (Kegeles et al., 2008). With wider use, it has been shown to worsen PD (Friedman et al., 2006; Wickremaratchi et al., 2006), and there are already reports of it causing parkinsonism (López-Torres et al., 2008), NMS (Chakraborty and Johnston, 2004; Hammerman et al., 2006; Palakurthi et al., 2007), acute dystonic reactions (Desarkar et al., 2006; Fountoulakis et al., 2006), and tardive dyskinesia (Zaidi and Faruqui, 2008; Abbasian and Power, 2009; Friedman, 2010).
Amisulpride
Amisulpride, a substituted benzamide, is a highly selective antagonist for dopamine D2 and D3 receptors in the limbic region, which would predict potent antipsychotic activity with a low potential to cause extrapyramidal symptoms (Lecrubier, 2000). It binds less tightly to the D2 receptor than do the typical antipsychotics (Seeman, 2002). Using the full range of recommended amisulpride dosage, striatal occupancies up to 90% can be measured (Vernaleken et al., 2004). It is not yet commercially available in the United States. It has already been reported to induce tardive oculogyric crisis (Mendhekar et al., 2010).
Adverse effects from second-generation antipsychotics
Aside from the D2-blocking effects described above, the five drugs in Table 19.7 have a number of other adverse effects. Sedation is a particular problem seen with each of them, but particularly clozapine and quetiapine. Metabolic side effects are now widely recognized with the second-generation antipsychotics, with weight gain a common problem (Leucht et al., 2009; Patel et al., 2009). Table 19.7 lists the common adverse effects of these drugs, along with haloperidol, for comparison.
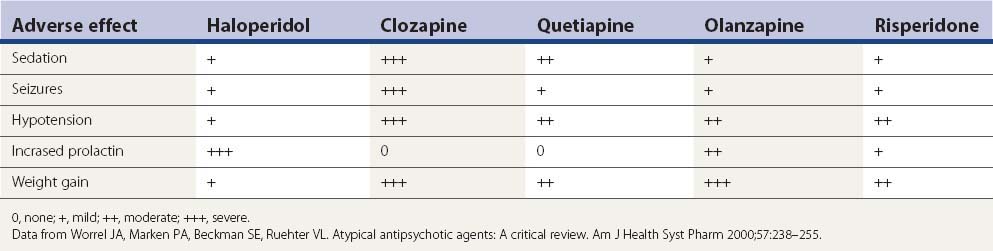
It is not likely that drug-induced tardive syndromes will disappear because use of both typical and atypical agents continues. In a survey in Lombardy (Italy), 35 363 individuals over the age of 65 were prescribed an antipsychotic prescription (2.18 subjects per 100 inhabitants, with two-thirds receiving first-generation agents) (Percudani et al., 2004). Although there may be a lower risk of developing these disorders with “atypicals,” these drugs can induce them (Table 19.8). It is even possible that if physicians consider “atypicals” safe to use, their use will increase, even in situations in which the risk from typicals might have precluded their use. In the United States, the prevalence of atypical antipsychotic use was found to be 267.1 per 100 000 subjects aged 19 years and younger and was more than twice as high for male patients as for female patients (Curtis et al., 2005).
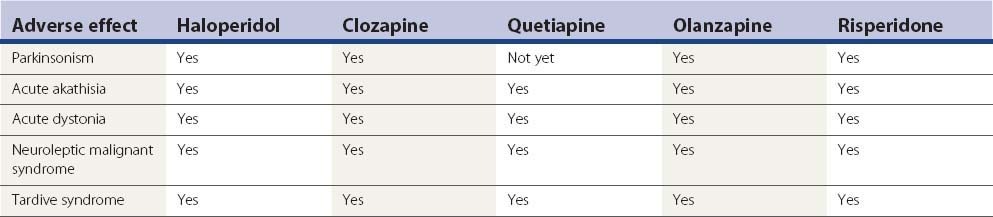
In a large US randomized controlled trial (the CATIE study) comparing second-generation antipsychotics (olanzapine, quetiapine, risperidone, perphenazine, or ziprasidone) with the first-generation perphenazine, only olanzapine had a slightly more favorable outcome of both the patients’ ability to stay on this drug for a longer duration and with slightly less drug-induced parkinsonism, akathisia, and tardive dyskinesia than all other drugs including perphenazine (Table 19.9); perphenazine fared as well as the other so-called ‘atypicals” (Lieberman et al., 2005). A similar outcome was seen in a smaller British study (CUtLASS) (Jones et al., 2006). This was a randomized controlled clinical trial involving 57 centers, with flexible dosing up to 4 capsules/day in 1460 schizophrenic subjects (after excluding 33 from one center, integrity concern). The primary outcome was time to discontinuation, with a maximum observation of 18 months. TD was present in 231 subjects at baseline, and these subjects were excluded from the perphenazine arm. The outcome had multiple comparison, requiring a P value ≤0.017 to be significant.
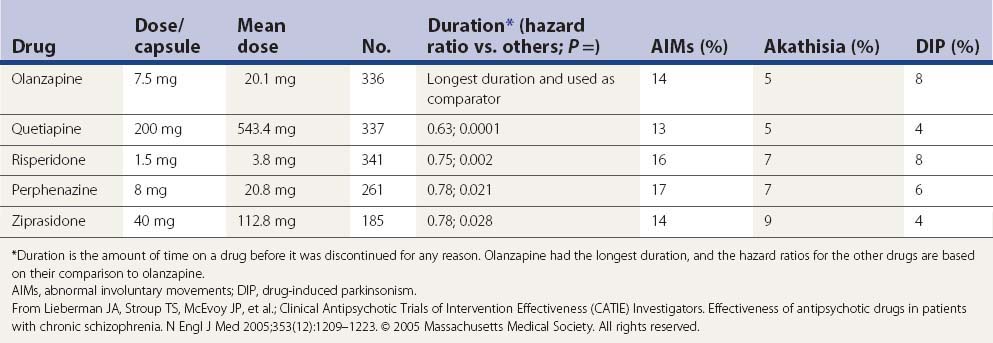
In a more detailed assessment of extrapyramidal side effects in the CATIE study, there were no significant differences in incidence or change in rating scales for parkinsonism, dystonia, akathisia, or tardive dyskinesia when comparing the second-generation antipsychotics with perphenazine or comparing between second-generation antipsychotics (Miller et al., 2008). Secondary analyses revealed greater rates of concomitant antiparkinsonism medication among individuals on risperidone and lower rates among individuals on quetiapine.
Metoclopramide
After the introduction of metoclopramide for gastrointestinal disorders, reports of acute dystonic reactions in children and tardive dyskinesia in adults began to appear (Cochlin, 1974; Kataria et al., 1978; Lavy et al., 1978). Today, metoclopramide has become a common cause of TD, and the legal profession has found such cases ripe for lawsuits. The annual incidence of TD from metoclopramide has been reported to be between 4.5% and 23% per year (Grimes, 1981; Ganzini et al., 1993), about the same as that for haloperidol ~5.7%/year (Tollefson et al., 1997; Beasley et al., 1999; Csernansky et al., 2002), but higher than those for risperidone ~1.8%/year (Lemmens et al., 1999; Davidson et al., 2000; Jeste et al., 2000; Csernansky et al., 2002) and olanzapine ~1.2%/year (Tollefson et al., 1997; Beasley et al., 1999). Because antipsychotic drugs have an affinity for brain neuromelanin (Seeman, 1988), Chen and colleagues (2010) examined the amount of metoclopramide and antipsychotic drugs binding to postmortem human substantia nigra. They found that in clinical conditions the amount of metoclopramide that would bind to nigra is much higher than the amount of raclopride, haloperidol, or olanzapine that would be expected to be bound and they suggest that this binding might explain the higher annual incidence of TD induced by metoclopramide.
Neurologic side effects of dopamine D2 receptor antagonists
To better understand the tardive syndromes, it is important to recognize the variety of other movement disorders that are induced by the dopamine receptor antagonists at different points in the course of treatment. These movement disorders are often lumped together as extrapyramidal syndromes, but the lumping often hinders the effort to sort out the clinical characteristics and pathophysiology of separate syndromes. It is better to subdivide them into their phenomenologic types (see Table 19.1). Movements that may be seen include acute dystonia, acute akathisia, parkinsonism, tardive syndromes, and NMS. Both dystonia and akathisia also occur as subtypes of tardive syndromes and both subtypes are discussed in more detail in the section on tardive syndromes.
Acute dystonia
The earliest abnormal involuntary movement to appear after initiation of dopamine receptor antagonist therapy is an acute dystonic reaction. In about half of the cases, this reaction occurs within 48 hours, and in 90% of cases, it occurs by 5 days after starting the therapy (Ayd, 1961; Garver et al., 1976). The reaction may occur after the first dose (Marsden et al., 1975).
Dystonic movements are sustained muscle contractions, frequently causing twisting and repetitive movements, or abnormal postures (Fahn, 1988). In a series of 3775 patients, Ayd (1961) found that acute dystonia is the least frequent side effect, affecting about 2–3% of patients, with young males being most susceptible. The incidence rate increases to beyond 50% with highly potent dopamine receptor blockers such as haloperidol (Boyer et al., 1987). In one prospective study, Aguilar and colleagues (1994) reported that 23 of 62 patients developed acute dystonia after haloperidol was introduced, that anticholinergic pretreatment significantly prevented this, and that younger age and severity of psychosis were risk factors. In the prospective study by Kondo and colleagues (1999), 20 (51.3%) of 39 patients placed on nemonapride had dystonic reactions, onset occurring within 3 days after the initiation of treatment in 90%. As in other series, the incidence of acute dystonia was significantly higher in males than in females (77.8% vs. 28.6%, P < 0.05), and younger males (≤30 years) had an extremely high incidence (91.7%). The incidence is much lower with the so-called atypical antipsychotics; Raja and Azzoni (2001) observed only 41 cases out of 1337 newly admitted patients treated with antipsychotics, which included 8 treated with risperidone, 1 with olanzapine, and 1 with quetiapine. A meta-analysis compared intramuscular injections of second-generation antipsychotics (SGAs) to haloperidol injections (Satterthwaite et al., 2008). SGAs were associated with a significantly lower risk of acute dystonia (relative risk = 0.19) and acute akathisia (relative risk = 0.25), compared with haloperidol alone.
All agents that block D2 receptors can induce acute dystonic reactions, including risperidone (Brody, 1996; Simpson and Lindenmayer, 1997) and clozapine (Kastrup et al., 1994). One child developed an acute dystonic reaction after ingestion of a dextromethorphan-containing cough syrup (Graudins and Fern, 1996). Dextromethorphan has several different known pharmacologic actions, but D2 receptor blockade is not one of them. A case of acute dystonia following abrupt withdrawal of bupropion (DA reuptake blocker) was reported (Wang et al., 2007). A case of acute dystonia following abrupt withdrawal of dexamphetamine in a patient taking risperidone was also reported (Keshen and Carandang, 2007). Serotonergic agents have also been reported to induce acute dystonic reactions (Lopez-Alemany et al., 1997; Madhusoodanan and Brenner, 1997; Olivera, 1997). The mechanism could relate to inadequate release of dopamine from the nerve terminals in the striatum owing to the inhibitory effect of serotonin on dopamine neurons in the substantia nigra pars compacta. The opioid σ1 and σ2 receptors have also been implicated (Matsumoto and Pouw, 2000).
Acute dystonic reactions most often affect the ocular muscles (oculogyric crisis), face, jaw, tongue, neck, and trunk, and less often the limbs. Oculogyric crisis has previously been noted to occur as a common feature of postencephalitic parkinsonism (Duvoisin and Yahr, 1965). A typical acute dystonic reaction may consist of head tilt backward or sideways with tongue protrusion and forced opening of the mouth, often with arching of trunk and ocular deviation upward or laterally (Rupniak et al., 1986). The forcefulness of the muscle contractions can be extremely severe and led to auto-amputation of the tongue in one patient (Pantanowitz and Berk, 1999).
Mazurek and Rosebush (1996) studied the timing in the development of an acute dystonic reaction in 200 patients who were taking a neuroleptic medication for the first time. The neuroleptic was given twice daily, and over 80% of the episodes of acute dystonia occurred between 12 noon and 11 p.m.
Reserpine and α-methylparatyrosine, which deplete presynaptic monoamines, have not been associated with acute dystonic reactions (Duvoisin, 1972; Marsden et al., 1975; Walinder et al., 1976). However, another dopamine depletor, tetrabenazine (TBZ), has been reported to induce acute dystonic reactions (Burke et al., 1985). One possible explanation for this difference is that in addition to depleting dopamine, TBZ blocks dopamine receptors (Reches et al., 1983).
It is important to mention the case described by Wolf (1973) of a patient who developed an oral dyskinesia while taking reserpine. Figure 1 in his paper clearly shows a dystonic phenomenon and not the complex, rapid, stereotypic movements of classic tardive dyskinesia. This development appeared too late after initiation of reserpine to be considered an acute dystonic reaction, however. Whether this could be an example of tardive dystonia is not clear, since the phenomenology of tardive dystonia is identical to that of naturally occurring primary dystonia; therefore, the patient could have had a coincidental case of spontaneous, idiopathic oromandibular dystonia (Fahn, 1984b). Thus, there is no absolute evidence that reserpine induces acute dystonic reactions, classic tardive dyskinesia, or tardive dystonia. In fact, symptoms of these three tardive syndromes can be suppressed by reserpine, and eventually reserpine can be withdrawn successfully in many patients without exacerbation of the symptoms (Fahn, 1983). Furthermore, the long time that reserpine has been available without a clear-cut case of tardive syndromes can be compared to the much shorter duration of use of metoclopramide, in which there are already cases of acute dystonic reactions, classic tardive dyskinesia, and tardive dystonia as a consequence of its use (Casteels-Van Daele et al., 1970; Gatrad, 1976; Pinder et al., 1976; Reasbeck and Hossenbocus, 1979; Miller and Jankovic, 1989; Lang, 1990).
Of 452 patients given high-dosage oral metoclopramide to control emesis, Kris and colleagues (1983) observed 14 who developed acute dystonic reactions. However, there was a distinct preponderance of the reactions occurring in children (6/22) compared to adults (8/430). Intravenous metoclopramide is more likely to induce it than oral administration (Pinder et al., 1976). In a study at a Veterans Administration hospital, comparing patients treated with metoclopramide and controls, the relative risk for TD was 1.67, and the relative risk for drug-induced parkinsonism was 4.0 (Ganzini et al., 1993).
The available biochemical explanations for the acute dystonic reaction are unsatisfactory, but several observations contribute to the understanding of the phenomena, relating it to dopamine, muscarinic, and sigma receptors. Acute neuroleptic administration produces sudden increase of dopamine release and increased turnover lasting for 24–48 hours after a single dose (O’Keefe et al., 1970; Marsden and Jenner, 1980). This effect is blocked by anticholinergics consistent with their efficacy in treatment of acute dystonic reactions (O’Keefe et al., 1970). Moreover, in baboons that were pretreated with reserpine and α-methylparatyrosine, which markedly reduces presynaptic dopamine concentration, haloperidol-induced acute dystonic reaction is abolished or greatly reduced (Meldrum et al., 1977). On the other hand, blockade of the postsynaptic receptor fades in about 12 hours after a single dose of antipsychotics, and supersensitivity of receptors begins to develop. Therefore, presynaptic dopaminergic excess in combination with the emerging supersensitive postsynaptic dopamine receptors could result in markedly increased striatal dopaminergic activity at about 20–40 hours after a neuroleptic dose. This period corresponds to the critical time for acute dystonic reactions in human subjects who were given a single dose of butaperazine (Garver et al., 1976). However, extrapolating the data from experimental animals to the clinical situation has its limitations including the fact that rats do not develop acute dystonic reactions. Further data on human cerebrospinal fluid (CSF) dopamine metabolites as an indicator of presynaptic function might prove to be of value in pursuing the hypothesis. Jeanjean and colleagues (1997) suggested that the σ2 receptors could be involved in the acute dystonic reaction. They found a correlation between the clinical incidence of neuroleptic-induced acute dystonia and binding affinity of drugs for the sigma receptor.
Another animal model of acute dystonia is the common marmoset that is treated with haloperidol (Fukuoka et al., 1997). But it takes at least 6 weeks of such treatment to develop this reaction. The dystonia subsides only to reappear when haloperidol treatment is restarted; other neuroleptics, including risperidone, can also make the dystonia reappear, but clozapine was without such an effect. In this animal model, the anticholinergic agent trihexyphenidyl inhibited the induction of acute dystonia.
In patients with acute dystonic reactions, symptoms can be relieved within minutes after parenteral anticholinergics or antihistaminics (Paulson, 1960; Waugh and Metts, 1960; Smith and Miller, 1961). Diphenhydramine 50 mg, benztropine mesylate or biperiden 1–2 mg is given intravenously and can be repeated if the effect is not seen in 30 minutes. Intravenous diazepam has also been shown to be effective and can be used as an alternative therapy (Korczyn and Goldberg, 1972; Gagrat et al., 1978; Rainer-Pope, 1979). If untreated, the majority of cases still resolve spontaneously in 12–48 hours after the last dose of the dopamine receptor antagonists. Dopamine receptor antagonists with high anticholinergic activities have low incidence rates of acute dystonic reactions (Swett, 1975). Therefore, prophylactic use of anticholinergics (Arana et al., 1988) and benztropine (Goff et al., 1991) has been studied and reported to be helpful in reducing the risk of acute dystonic reactions, especially in young patients on high-potency drugs. Three cases of recurrent episodes of acute dystonia and oculogyric crises despite withdrawal of the offending DRBAs (haloperidol in two cases, metoclopramide in one) have been reported, responding to anticholinergics each time (Schneider et al., 2009). The response of acute dystonic reactions to anticholinergics is so characteristic that it is difficult to explain the report of a few cases apparently due to amitriptyline, which has considerable anticholinergic activity and no known DRBA activity (Ornadel et al., 1992).
A case of an acute dystonic reaction occurring in an elderly person with bipolar disorder taking the serotonin uptake inhibitor paroxetine has been reported (Arnone et al., 2002). Speculation about the risk due to previous exposure to neuroleptics was raised. This class of drug can reduce firing rate of nigral dopaminergic neurons owing to their inhibition by serotonin.


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


