Tiagabine
Reetta Kälviäinen
Introduction
Tiagabine (Gabitril) (Fig. 1) is a selective γ-aminobutyric acid (GABA) reuptake inhibitor (SGRI), which increases synaptic GABA availability via inhibition of the GAT-1 GABA transporter on presynaptic neurons and glial cells.10 It is structurally related to nipecotic acid, but has an improved ability to cross the blood–brain barrier. Tiagabine has been approved for add-on therapy in patients with refractory partial seizures with or without secondary generalization in approximately 30 countries.
Mechanism of Action
Potentiation of GABA is regarded as one of the most important mechanisms of action for novel antiepileptic agents. GABA uptake inhibitors represent a new class of antiepileptic drugs (AEDs), of which tiagabine was the first to be introduced into clinical practice. Tiagabine prevents GABA uptake by inhibiting selectively the GAT-1 GABA transporter, with little or no activity on GAT-2, GAT-3, or BGT-1, which are the GABA transporters responsible for the uptake of the neurotransmitter into neurons and glial cells after synaptic release.10,60 Tiagabine’s affinity for inhibiting glial GABA uptake is 2.5-fold greater than that for neuronal uptake.11 Tiagabine is not a substrate for the GABA uptake carrier and is therefore unlikely to act as a false transmitter at GABAergic neurons.11 Blockade of GABA uptake temporarily sustains levels of endogenously released GABA in the synapse.60 This is the only known mechanism of tiagabine action.
Although both tiagabine and vigabatrin act by enhancing GABA neurotransmission in the central nervous system (CNS), preclinical data show that vigabatrin and tiagabine have different pharmacologic profiles and different mechanisms of action at the cellular level.55 Tiagabine prolongs the duration, but not the magnitude, of the peak inhibitory postsynaptic current, consistent with temporarily sustained levels of endogenously released GABA in the synapse.49 By contrast, vigabatrin inhibits presynaptic GABA degradation by selective, enzyme-activated irreversible blockade of the mitochondrial enzyme GABA transaminase, and thus it induces a persistent fivefold increase in whole brain GABA concentration, and also high concentrations in the retina.56 Tiagabine does not induce the widespread increase in total brain GABA concentrations that accompanies GABA-T inhibition. Moreover, viga-batrin seems to accumulate in the retina, whereas tiagabine does not.56
Experimental Studies
The anticonvulsant action of tiagabine has been studied in animal models of epilepsy induced by electrical, chemical, and sensory stimuli, and in genetic models of epilepsy. When administered intraperitoneally to amygdala-kindled rats, tiagabine attenuates the expression of secondarily generalized seizures and completely blocks the expression of partial seizures.20 Tiagabine also suppresses amygdala kindling–induced epileptogenesis in a dose-dependent manner in the rat.21
Intraperitoneal tiagabine was shown to be an active anticonvulsant in experimental studies by protecting against audiogenic and methyl-6,7-dimethoxy-4-ethyl-β-carboline-3-carboxylate- and pentylenetetrazol (PTZ)-induced tonic or clonic seizures in mice and PTZ-induced tonic or clonic seizures in rats.46 However, tiagabine did not protect against maximal electroshock-induced tonic seizures in mice or rats.20,46 Tiagabine did not prevent tonic or clonic seizures induced by the potassium channel antagonists dendrotoxin or 4-aminopyridine in mice.16,64
In common with GABA agonists, tiagabine may enhance the occurrence of spike-and-wave discharges in animal models of nonconvulsive epilepsy. In WAG/Rij rats, a genetic model of generalized nonconvulsive absence epilepsy, spike-and-wave discharges were increased by tiagabine 3 and 10 mg/kg administered intraperitoneally, but not by a 1 mg/kg dose.15 Walton et al.63 reported that tiagabine was effective in treating status epilepticus (SE) in cobalt-lesioned rats. At doses ≥5 mg/kg intraperitoneally, it produced rhythmic high-voltage discharges. At even higher doses, a similar pattern could be produced in normal rats as well. Exacerbation of absence seizures has been found also in rats with genetic absence epilepsy (GAERS) and in the lethargic mouse model.29
Tiagabine reduced both seizure-induced damage to pyramidal cells in the hippocampus and impairment of spatial memory associated with hippocampal damage in the perforant pathway stimulation model of SE in the rat.28 Neuronal cell death was also reduced by tiagabine in the hippocampus of gerbils subjected to cerebral ischaemia30 and in the rat cerebral ischaemia model of delayed pyramidal cell death.31
Pharmacokinetics
Tiagabine is rapidly and nearly completely absorbed after oral administration, with peak concentrations seen within 30 to 90 minutes of dosing.27 Food delays the time to peak concentration from a mean of 0.9 to a mean of 2.6 hours, but does not change the total quantity absorbed. Because tiagabine has a short elimination half-life, the smoother absorption produced by concomitant intake of food helps in reducing excessive fluctuations in plasma drug levels during the dosing interval and, for this reason, it is recommended that the drug be taken at meal times, preferably at the end of the meal.
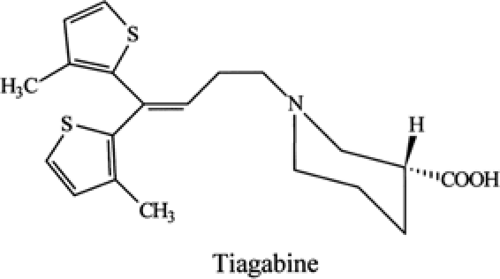 FIGURE 1. Chemical structure of tiagabine. |
Table 1 Efficacy of tiagabine as add-on therapy in partial epilepsy | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Protein binding is high at 96%, but tiagabine does not displace highly protein-bound drugs such as phenytoin and valproic acid from their binding sites. The volume of distribution is approximately 1 L/kg. Tiagabine is widely metabolized in humans, mainly by isoform CYP3A4 of the cytochrome P450 family. Less than 1% is excreted unchanged in the urine, and no active metabolites have been identified.45
Tiagabine pharmacokinetics are linear at doses up to 80 mg/day.58 There is no evidence that tiagabine either causes induction or inhibition of cytochrome P450 enzymes.27 Consequently, tiagabine has shown no action on hepatic metabolism, which would be expected to alter the pharmacokinetics of cimetidine, carbamazepine, digoxin, erythromycin, oral contraceptives, phenytoin, theophylline, valproate, and warfarin.12 However, enzyme-inducing AEDs such as carbamazepine, phenytoin, phenobarbital, or primidone increase the hepatic clearance of tiagabine when given in combination.32 The plasma half-life, which is normally 5 to 9 hours,27 is reduced to 2 to 3 hours in combination with enzyme-inducing drugs.
Clinical Efficacy
Evidence from Randomized Controlled Trials
Tiagabine has proven effective as add-on therapy in patients with refractory partial seizures with or without secondary generalization. The primary clinical evidence for this efficacy is based on five controlled add-on trials in adults with epilepsy unsatisfactorily controlled with current AEDs (Table 1).
The first phase II multicenter trials were two small, placebo-controlled cross-over studies. In an initial titration period lasting up to 8 weeks, patients started with a tiagabine dose of 8 mg/day, and the dose was titrated either to reduce seizures sufficiently or to produce unacceptable adverse events. Patients then entered a 4-week fixed-dose period on the dose attained in titration. The maximal dose allowed in the first study was 52 mg/day.48 Patients were eligible to enter the double-blind cross-over phase if their seizure frequency had been reduced by at least 25% during the fixed-dose period. In this two-period crossover study, patients were randomized to placebo/tiagabine or their previously determined dose of tiagabine/placebo, remaining on each of these two regimens for 7 weeks. The 7-week treatment periods were separated by a 3-week washout period. The median daily dose of tiagabine in the double-blind phase was 32 mg/day. Of the total of 42 patients who contributed data for both periods of the crossover phase, 26% of those with complex partial seizures and 63% with secondarily generalized tonic–clonic seizures (n = 27) experienced a reduction of at least 50% in seizure frequency during the tiagabine period compared with the placebo period. The median seizure rate during the tiagabine treatment period was significantly lower than during the placebo treatment period for complex partial seizures (p = 0.05) and secondarily generalized tonic–clonic seizures (p = 0.009).
The second phase II study used the same design but allowed a maximal dose of 64 mg/day.18 The intent-to-treat group comprised 36 patients who received a mean total daily dose of 46 mg in the tiagabine treatment periods. Tiagabine was significantly better than placebo in reducing all partial seizures (p = 0.002), complex partial seizures (p < 0.001), and partial seizures with secondary generalization (p = 0.030). A total of 46% of patients with complex partial seizures had at least a 50% reduction in weekly seizure rates.
Altogether 769 patients took part in the three multicenter, parallel-group, double-blind add-on studies in which tiagabine was compared with placebo—a dose-response study, a dose-frequency study, and a three-times-a-day (t.i. d.) dosing study.35,50,62 The dose-ranging multicenter study in the United States had a fixed-dose, placebo-controlled parallel-group design (n = 297)62. During a 4-week period, tiagabine-treated patients were given increasing doses until the dose level to which they had been randomized was reached (16, 32, or 56 mg/day, divided in four equal doses). The patients then remained on a fixed dose for 12 weeks of double-blind treatment. Median decreases in 4-week complex partial seizure frequency for 32 mg (–2.2) and 56 mg (–2.8) tiagabine groups were significantly greater than for the placebo (–0.7) group (P = 0.03 and P < 0.03, respectively); 20% and 29% of patients in the 32- and 56-mg groups had a 50% or greater reduction in the
frequency of complex partial seizures compared with 4% in the placebo group (P = 0.002 and P < 0.001, respectively).
frequency of complex partial seizures compared with 4% in the placebo group (P = 0.002 and P < 0.001, respectively).
The dose-frequency study was also a randomized, double-blind, placebo-controlled U.S. multicenter study with a parallel-group, add-on design (n = 318).50 The study lasted for 24 weeks and consisted of an 8-week baseline, a 12-week double-blind treatment phase, and a 4-week termination period. During the first month of treatment, doses were increased weekly to 32 mg/day. The treatment groups were placebo, 16 mg tiagabine twice a day (b.i.d.) and 8 mg tiagabine four times a day. The median changes in 4-week complex partial seizure rates were –1.6 (p = 0.055) for the 16 mg twice-a-day group and –1.2 (p <0.05) for the 8 mg four-times-a-day group, versus –0.2 for placebo. Statistically significant differences between placebo and two tiagabine groups occurred in the proportion of patients experiencing >50% rate reduction for complex partial, simple partial, and all partial seizure rates.
The thrice-daily dosing study was a Northern-European multicenter parallel-group study that compared a dose of 30 mg/day tiagabine with placebo as add-on therapy (n = 154).35 The study included 12-week baseline, an 18-week double-blind treatment phase, and a 4-week termination period. The median change from baseline in complex partial seizure rates was –1.3 for patients on tiagabine, whereas placebo patients had a median increase of 0.1 in complex partial seizure rates (p <0.05). Tiagabine was significantly more effective than placebo in patients with simple partial seizures with respect to the proportion of patients achieving a seizure reduction of at least 50% (21% vs. 6%; p <0.05).
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


