Brain CT scan showing an old infarct in the left middle cerebral artery territory.
As he was leaving the CT-scan room, he had brief involuntary movements of the right hemibody, with twitching of the right side of the face, and right gaze deviation. The involuntary movements lasted for approximately 20 seconds. Brain magnetic resonance imaging (MRI) with diffusion-weighted imaging (DWI) and apparent diffusion coefficient (ADC) sequences showed no evidence of acute ischemic lesions.
Diagnostic reasoning
In the emergency department, physicians have to decide promptly what is the most probable diagnosis and what the next action will be. When our patient arrived at the emergency department, his neurologic examination and presentation were compatible with an acute ischemic stroke of the left MCA territory. He had sudden onset of focal neurologic deficits and had multiple vascular risk factors. As the patient was last seen without symptoms within 50 minutes before arriving at the emergency department, the patient was well within the 4.5 hour time window from symptom onset or from last seen well to administer intravenous thrombolysis with recombinant tissue plasminogen activator (tPA). The CT was also unremarkable for acute ischemic stroke and excluded other disorders that could mimic an acute ischemic stroke such as primary central nervous system (CNS) tumors, metastasis, brain abscess, or hemorrhages.
However, the adventitious movements experienced by the patient as he was leaving the CT-scan room raised concerns for a possible seizure secondary to an acute ischemic stroke, or a seizure with a Todd’s paralysis since the very onset. Partial or secondary generalized seizures may occur in the acute phase of ischemic stroke. Initially described in the nineteenth century by Robert Bentley Todd, Todd’s paralysis refers to a post-seizure event, defined as a transient weakness and depression of motor ability lasting hours to days. It usually affects one or more limbs and follows a focal seizure, or more rarely a generalized tonic-clonic seizure [1]. Sometimes, there may be other neurologic signs such as aphasia, neglect, or psychosis depending on the epileptic focus and surrounding focal areas involved. Therefore, features of post-seizure Todd’s paralysis may be similar to an acute ischemic stroke. Several clinical series present unwitnessed or unrecognized seizures with the post-ictal state misdiagnosed as stroke being the most common stroke mimic. Some small series suggest that patients with structural lesions, such as those with previous brain infarcts and the elderly, may be more likely to have transient focal weakness following a seizure. Although there is no consensus regarding the pathophysiology of Todd’s paralysis, hypotheses regarding its pathogenesis include: neuronal desensitization, neurotransmitter depletion, active suppression and exhaustive neuronal firing, and localized cerebral hypoperfusion resulting from motor cortex exhaustion.
A detailed description of the onset of symptoms is critical to distinguish between Todd’s paralysis and an acute ischemic stroke. However, patients are often unable to report onset of symptoms either because they are aphasic or unaware of their deficits. In these cases, accompanying family members may provide valuable information. However, in our patient, symptom onset was not witnessed, and doubts regarding the precise diagnosis remained. This diagnostic uncertainty has a clear repercussion in management, as interventional stroke therapies have potential serious side effects such as intracranial bleeding. Also, although patients with seizures at stroke onset were excluded from thrombolytic trials due to the possibility of confusion with Todd’s paralysis, case series suggest that thrombolysis could be useful if there is evidence of a new ischemic stroke. Therefore, it is currently recommended by the European Stroke Organisation that intravenous tPA may be used in patients with seizures at stroke onset, if the neurologic deficit is related to acute cerebral ischemia (Class IV, Good Clinical Practice – GCP) [2]. Guidelines from the American Heart Association/American Stroke Association (AHA/ASA) [3] state that “intravenous tPA is reasonable in patients with a seizure at the time of onset of stroke, if evidence suggests that residual impairments are secondary to stroke and not a postictal phenomenon” (Class IIa, Level of Evidence C).
The elderly have a high incidence of epilepsy that may be related to cerebrovascular disease [4]. In our patient, there were subtle symptoms in favor of a seizure with Todd’s paralysis such as his sleepiness, his neurologic improvement, and the past history of a cortical infarct.
However, our patient was found lying on the floor with an acute focal neurologic deficit. He had multiple vascular risk factors including hypertension, dyslipidemia, and a previous stroke, all of which could have been in favor of a new stroke. However, our patient did not have a prior history of seizures.
In these situations, MRI with DWI/ADC sequences can be useful to exclude the presence of acute cerebral lesions not yet detectable with a CT scan. DWI/MRI is the most useful exam to differentiate between an acute ischemic stroke and stroke mimics. Of note, there are reports of hyperintensities on brain DWI/MRI in Todd’s postepileptic paralysis described as gyriform cortical hyperintensities that do not necessarily reflect ongoing seizure activity. These image changes are not associated with a particular vascular distribution, and rather reflect the epileptic foci and surrounding tissue.
Brain CT angiography may be an indirect useful modality in differentiating Todd’s paralysis from early seizure and ischemia by detection of intracranial occlusions, which would favor a diagnosis of an ischemic lesion [5]. Electroencephalography (EEG) does not help in differentiating these entities, as focal slowing and epileptiform activity can occur in both acute ischemia and in the post-seizure period.
Tip
An adequate clinical history that properly characterizes time of symptom onset is critical to distinguish TIA or stroke from other pathologies that can also present with focal neurologic deficits.
Case 2. Headache and hemiparesis
Case description
A 21-year-old woman noticed that upon standing up from a chair, her left side became weak and numb. One hour later, she had a throbbing left-sided headache and felt nauseous. She had a history of recurring unilateral throbbing headaches of moderate intensity accompanied by nausea, photophobia, and sonophobia. Headaches were severe enough to interfere with her daily activities. One year earlier, she had a similar episode of headache, preceded by a feeling of heaviness of the right arm which subsided in a few hours. Her sister and mother had a similar history of headaches and occasional weakness. On admission, her blood pressure was 105/60 mmHg. When she arrived at the emergency department she had a left central facial paresis, a left hemiparesis (Medical Research Council grade 4), and left hemisensory loss. Blood glucose was 110 mg/dL. Brain MRI was unremarkable. The headache resolved within 3 hours, but her left hemiparesis persisted for 5 more hours.
Diagnostic reasoning
The young age of our patient and the lack of traditional vascular risk factors raised the possibility of an alternative diagnosis to a TIA. TIAs are rare in young patients without vascular risk factors. The presence of recurrent transient neurologic deficits followed by an evanescent headache led to the diagnosis of migraine with aura. Migraines usually start in the first or second decades of life. Our patient had a past history of pulsatile, moderate intensity, unilateral headaches with nausea and photophobia that suggest a diagnosis of migraines [6]. Migraine is a common disorder that can be accompanied by aura in about a third of patients. Migraines with aura can present with neurologic symptoms resembling a TIA or stroke. An aura is defined as transient focal neurologic symptoms that usually precede, or sometimes accompany, the headache. The symptoms usually last minutes and are fully reversible, and are usually followed by a headache and other associated migrainous symptoms [7]. However, the headache may sometimes be absent masking the correct diagnosis. The typical migraine aura is most commonly visual, but it may also be characterized by sensory, speech, or language difficulties. Visual symptoms may be characterized by positive features such as zigzag lines or flickering spots with or without negative features such as scotomata. Basilar migraine may present with double vision, unsteadiness, fainting, or losing consciousness. Retinal or ophthalmic migraine typically affects only one eye. Whenever the aura includes weakness as a symptom, it is classified as hemiplegic migraine, as in our patient. Distinguishing motor from sensory auras can be challenging at times. The motor aura should be a clearly characterized motor deficit such as weakness with difficulty moving one hand, arm, or leg. Some patients with a sensory aura may report dropping objects. A population-based epidemiological survey found a prevalence of 0.005% for hemiplegic migraine [8]. Patients with hemiplegic migraine may, in addition to motor aura, have any of the aura symptoms of migraines with aura or basilar migraine [7]. Most patients also have attacks of migraines with typical aura – without weakness. The most common accompanying aura symptom besides weakness in these patients is sensory. The typical sensory aura is characterized by tingling involving one of the digits that gradually progresses to involve other digits, up to the arm, and then affects the face, tongue, and later the body and leg. The different aura symptoms progress slowly over 20–30 minutes and occur successively, mainly in the following order: visual, sensory, motor, aphasic, and basilar disturbances.
Migraine aura is considered to be caused by cortical spreading depression, which is characterized by a brief neuronal excitation that initiates a depolarization wave that moves across the cortex followed by a prolonged inhibition of neuronal activity [9]. Hemiplegic migraine can occur as a sporadic or a familial disorder. Patients with sporadic hemiplegic migraine lack a family history of at least one affected first-degree or second-degree relative. The sporadic and hemiplegic migraine forms have a similar prevalence of 0.002–0.003%. Familial hemiplegic migraine (FHM) is dominantly inherited. The presence of similar symptoms in both her sister and mother suggest this type of inheritance in our patient. FHM1 is caused by mutations in the CACNA1A gene located in chromosome 19, FHM2 by mutations in the ATP1A2 gene located in chromosome 1, and FHM3 by mutations in the SCN1A gene located in chromosome 2. All the involved genes take part in ion transport. Both sporadic and familial forms of hemiplegic migraine have similar clinical presentations. The mean frequency of episodes is approximately three per year; the frequency and severity tend to decrease with advancing age.
Imaging and cerebrospinal fluid (CSF) studies done during or after an episode of migraine are unremarkable, except in FHM1 where cerebellar atrophy may be present.
Headache is a common feature of acute ischemic stroke. Twenty-seven percent of patients experience a headache at stroke onset. Sometimes, headache in patients with stroke may point to a cervical artery dissection as the cause. There is also a specific type of stroke related to migraine – migrainous infarction. In migrainous infarction the symptoms associated with the typical aura are not fully reversible. The International Classification of Headache Disorders, 3rd Edition (ICHD-3) beta version defines migrainous infarction as one or more otherwise typical auras persisting beyond one hour with neuroimaging confirmation of an ischemic infarction in the affected territory [6]. Meningitis and intracranial venous thrombosis may also present with headaches and focal neurologic deficits, but usually other diagnostic clues are present.
Case 3. Sudden memory impairment
Case description
A 65-year-old woman was admitted to hospital due to a sudden onset of memory impairment. Early in the morning, she telephoned her husband telling him she had vertigo and was not feeling well. Five minutes later, when her husband found her, she made him repeat questions: Where am I? What happened? What am I doing here? What day is this? Although he answered appropriately, she did not seem to remember his answers, as she repeated the same questions over and over again. She also did not remember what she had done the day before. This episode lasted 5 hours, with a gradual improvement in her ability to recall information. During the episode, she never lost contact with her husband. He did not notice any involuntary movements or automatisms. When she was seen at the emergency department, she was able to recall new information. However, she could not remember what had happened early in the day. She had a past history of hypertension and was under treatment with lisinopril. There was no history of recent head trauma. She did not take any drugs or new medications. On admission, she was afebrile. Blood pressure was 113/70 mmHg. Neurologic examination was unremarkable, including her ability to recall new information, tested by the three-word test/word list and evocation of recent events. Blood glucose was 120 mg/dL. Brain MRI with DWI and ADC sequences were unremarkable. EEG showed focal slowing or epileptiform activity. Follow-up brain MRI with diffusion sequence performed 3 days later showed a rounded image restriction in the CA1 segment of the right hippocampus (Figure 1.2). She did not have further symptoms.
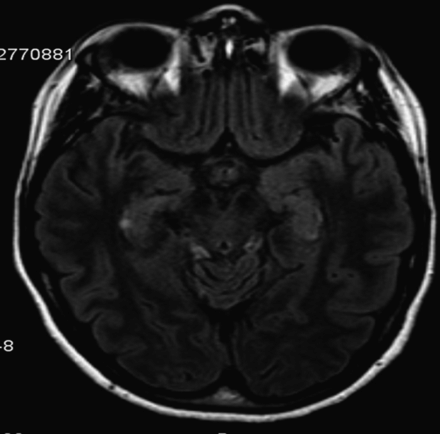
Brain MRI showing a rounded image in the CA1 segment of the right hippocampus.
Diagnostic considerations
This sudden impairment of memory is consistent with transient global amnesia (TGA). The incidence of TGA ranges between 3 and 8 per 100 000 people per year [10]. TGA is characterized by sudden onset of anterograde and retrograde amnesia lasting for up to 24 hours, but usually lasting substantially less. There should be no loss of personal identity, personality, language, or visuospatial functions during the amnestic episode. Also, no other neurologic deficits, recent head trauma, or signs of a seizure should be present. After the episode, anterograde memory returns to normal, but the patient may never remember what happened during the period of amnesia. TGA most commonly occurs in patients in their sixth or seventh decade of life. Headaches, dizziness, or nausea may be present during an episode of TGA. Strenuous physical activities or strong emotional events may antedate onset of symptoms [11]. Neuroimaging studies following an acute TGA event show transient perturbation of specific hippocampal circuits involved in memory processing. Focal diffusion lesions can be selectively detected in the CA1 field of the hippocampal cornu ammonis on brain MRI with DWI/ADC when done approximately 72 hours after symptom onset.
Although migraine, focal ischemia, venous flow abnormalities, and epileptic phenomena have been implicated in the pathophysiology of TGA, the factors triggering these unique events remain unknown. Recent data suggest that the vulnerability of the CA1 neurons to metabolic stress plays an important role in the pathophysiological cascade, leading to an impairment of hippocampal function during TGA [12].
There are cases of transient amnesia due to focal seizure activity also known as transient epileptic amnesia. Transient epileptic amnesia has clinical presentation similar to TGA episodes and tends to occur in the morning hours, but it can be distinguished by the shorter duration and repeated amnestic periods.
TIAs or strokes with memory impairment are rare. Every region of the limbic system involved in memory processing may be damaged by strokes, but very rarely in isolation. The combination of amnesia with other acute associated neurologic deficits often leads to the suspicion of a cerebrovascular event in these patients [13].
Case 4. Focal neurologic deficits due to metabolic disturbances
Case description
A 60-year-old woman was admitted to hospital half an hour after her husband noticed increased somnolence, confusion, and decreased strength in her left arm and leg. She had taken her medications during the morning and had gone jogging. Two hours later, her husband found her to be somnolent, with confused speech and difficulty moving her left arm and leg. She had a past history of diabetes mellitus type 2 and ischemic heart disease; she was on metformin, glibenclamide, aspirin, and bisoprolol. Upon admission, neurologic examination showed impaired alertness, confusion, and left hemiparesis (Medical Research Council grade 3). Blood glucose level was 50 mg/dL. She immediately received intravenous 10% glucose. Brain CT scan was unremarkable. Approximately 20 minutes following correction of her blood glucose levels, she started to improve, and made a complete recovery 5 hours after symptom onset. It was later determined that the patient had taken a higher dose of glibenclamide.
Diagnostic considerations
Hypoglycemia and less often hyperglycemia (blood glucose concentration >400 mg/dL) can cause focal neurologic deficits that can mimic a TIA or stroke [14]. Hypoglycemia is a very important differential diagnosis of TIA and stroke. Generally defined as a blood glucose level <70 mg/dL (3.9 mmol/L), it is more common among patients with diabetes mellitus. However, any patient can have hypoglycemia. Causes of hypoglycemia include medications (insulin or sulfonylureas), exercise, fasting, alcohol, insulin secreting tumors, and endocrine disorders such as Addison’s disease. In our patient, a personal history of diabetes mellitus and antidiabetic medications namely with sulfonylureas (glibenclamide) should lead to the suspicion of a broad differential diagnosis including hypoglycemia. Glucose is an obligate metabolic fuel for the brain under physiologic conditions, and it is essential for brain metabolism as the brain cannot synthesize glucose or store more than a few minutes’ supply as glycogen. Therefore, the brain needs a continuous arterial supply of glucose. As the plasma glucose concentration falls, blood-to-brain glucose transport becomes insufficient to support brain energy metabolism and function. If hypoglycemia is severe and prolonged and remains untreated, it can lead to a life-threatening situation. Acute effects of hypoglycemia are primarily neurologic. Symptoms of hypoglycemia are initially related to catecholamine release, and later, if untreated, due to neuroglycopenia. Autonomic symptoms include tachycardia, diaphoresis, tremor, anxiety, and hunger. These symptoms are important warnings; however, they may be lacking, for example, among diabetic patients with autonomic insufficiency or in patients on beta-blockers like our patient. Beta-blockers may mask the sympathetic nervous system manifestations of hypoglycemia, and therefore, patients may manifest only symptoms of neuroglycopenia. Neuroglycopenic symptoms are usually present when levels of blood glucose are <50 mg/dL (<2.8 mmol/L). However, these thresholds are dynamic, and in patients with poorly controlled diabetes mellitus, these thresholds are shifted to higher blood glucose concentrations. Neuroglycopenic symptoms include impairment of consciousness that can progress to coma if untreated, confusion, abnormal behaviors, seizures, headaches, and focal neurologic symptoms [15]. Therefore, measurement of blood glucose levels should be part of the initial evaluation of all patients presenting with focal neurologic deficits. Focal neurologic deficits in hypoglycemia may include aphasia, homonymous hemianopsia, hemisensory deficits, hemiparesis, unilateral hyperreflexia, and Babinski sign(s). Both the American Heart Association (AHA) and the European Stroke Organisation (ESO) recommend determination of blood glucose concentration in their guidelines for the evaluation of stroke patients [1,2]. When hypoglycemia is detected, treatment should be started as soon as possible. Both intravenous dextrose and infusion of 10–20% glucose can be used to correct hypoglycemia. Thiamine, 100 mg intravenously or intramuscularly, is given to patients with alcohol dependence to prevent Wernicke’s encephalopathy before administering glucose. If intravenous therapy is not possible, subcutaneous or intramuscular glucagon may be used. For non-hypoglycemic patients, excessive dextrose-containing fluids have the potential to exacerbate cerebral injury. Therefore, normal saline is more appropriate if rehydration is required. Of note, there can be a delay of hours to days between correction of blood glucose concentration and improvement of neuroglycopenic symptoms. If abnormalities persist longer than 30 minutes following glucose administration and hypoglycemia has not recurred, other causes should be investigated with brain imaging and appropriate laboratory evaluation.
Imaging abnormalities in patients with hypoglycemia are uncommon but very variable, weakly associated with the neurologic deficits, and about a fifth may mimic an acute ischemic stroke. Diffuse and extensive injury observed on DWI with MRI predicts a poor neurologic outcome in patients with hypoglycemic injuries [16].
Other metabolic disturbances that may account for focal neurologic deficits include hyponatremia, hypernatremia [17], and hepatic encephalopathy [18].
Related posts:
 Recognizing cerebrovascular etiologies of “thunderclap†headaches
Choosing the right patients for carotid artery procedural interventions
Transient ischemic attacks (TIAs) – an underrecognized and undertreated disorder
The diagnosis and overdiagnosis of cerebral vasculitis
Dilemmas in endovascular stroke therapy
Underdiagnosis of reversible cerebral vasoconstriction syndromes
Recognizing cerebrovascular etiologies of “thunderclap†headaches
Choosing the right patients for carotid artery procedural interventions
Transient ischemic attacks (TIAs) – an underrecognized and undertreated disorder
The diagnosis and overdiagnosis of cerebral vasculitis
Dilemmas in endovascular stroke therapy
Underdiagnosis of reversible cerebral vasoconstriction syndromes
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


