Patient Involvement
Involvement of the patient is helpful, not only in making initial decisions which are therefore more likely to be followed but also in monitoring the course of the disease. Home BP readings should always be recommended, preferably taken by the patient or sometimes by other caregivers. Moreover, as noted in Chapter 2, responses to therapy are more closely related to out-of-office measurements than office readings.
Intensity of Therapy
The rapidity of reaching goal is now in question since too fast a course may cause intolerable symptoms, but too slow may expose high-risk patients to immediate dangers. The benefit of more rapid control was graphically shown in the Valsartan Antihypertensive Long-term Use Evaluation (VALUE) trial, wherein the quicker response over the first 3 to 6 months to the CCB amlodipine than to the angiotensin II receptor blocker (ARB) valsartan, provided greater protection against heart attacks and strokes (Julius et al., 2004). The patients in VALUE were all at high risk for cardiovascular disease, so it still seems appropriate to “start low and go low” for most, particularly the elderly with systolic hypertension. For those with higher levels of BP and, even more so, with higher overall risk, a “higher and faster” approach may be more appropriate.
Timing of Dosing
The time of day to take one-a-day antihypertensive medications needs to be more carefully considered. Early morning has usually been recommended, but there are two potential problems: First, the pills may not exert a full 24-hour effect; second, an even greater effect may be needed in the early morning, before today’s therapy has kicked in, to keep the pressure from surging in the immediate postarising time, thereby contributing to the “morning surge” of cardiovascular catastrophes.
The solution for the first problem is twofold: First, ensure full 24-hour control by having the patient measure early morning BP at home; second, choose intrinsically long-acting medications, e.g., metoprolol XL rather than atenolol, trandolapril rather than enalapril, and amlodipine rather than felodipine.
The solution for the second problem seems as obvious: i.e., take medications later in the day or even at bedtime. A group of investigators from Vigo, Spain, have published the results of multiple studies in hypertensives, including some with diabetes or chronic kidney disease, that show improved BP control and cardiovascular outcomes with bedtime rather than morning dosing (Hermida et al., 2011).
In a Cochrane Review of the 21 RCTs published between 1978 to 2009 that compared morning versus evening dosing regimens, the authors concluded, “There were no significant differences in overall adverse events and withdrawals due to adverse events among the evening versus morning dosage regimens. In terms of BP lowering efficacy, for 24-hour SBP and diastolic blood pressure (DBP), the data suggest that better blood pressure control was achieved with bedtime dosing than morning administration of antihypertensive medication, the significance of which is not known” (Zhao et al., 2011).
Dealing with Side Effects
Some medications are easier to take than others, but some patients cannot seem to take any. Such patients with nonspecific intolerance to multiple antihypertensive drugs almost always have underlying psychological morbidity, often manifested as recurrent hyperventilation, panic attacks, generalized anxiety, or depression (Davies et al., 2003). As noted before, the labeling of hypertension can induce adverse psychological effects (Spruill et al., 2012). Fortunately, the use of currently available drugs slows cognitive decline and may prevent dementia (Marpillat et al., 2013), but some aspects of the quality of life (QOL) may be worse in hypertensives under drug treatment than equally hypertensive patients on no medication (Trevisol et al., 2012).
Follow-Up Visits
To achieve and maintain target BP with the lowest possible dosage of medication requires ongoing patient follow-up, preferably with home blood pressure monitoring (BPM), and may involve multiple dosage adjustments. Most patients should be seen within 1 to 2 months after the initiation of therapy to determine the adequacy of BP control, the degree of patient cooperation in taking pills, the need for more therapy, and the presence of adverse effects. Once the BP is stabilized, follow-up at 3- to 6-month intervals is generally appropriate. In most patients, particularly the elderly and patients with orthostatic symptoms, monitoring should include BP measurement in the supine position and after standing for up to 5 minutes, to recognize postural hypotension.
SPECIFICS ABOUT ANTIHYPERTENSIVE DRUGS
The modern era of antihypertensive therapy began only about 55 years ago with the pioneering work of Ed Freis in the U.S. and Horace Smirk in New Zealand (Piepho & Beal, 2000). Since then, a large panoply of drugs have been developed, as listed in Table 7-2. We will consider the drugs in the order shown in Table 7-2.
TABLE 7-2 Antihypertensive Drugs Available in the U.S. (as of 2014)
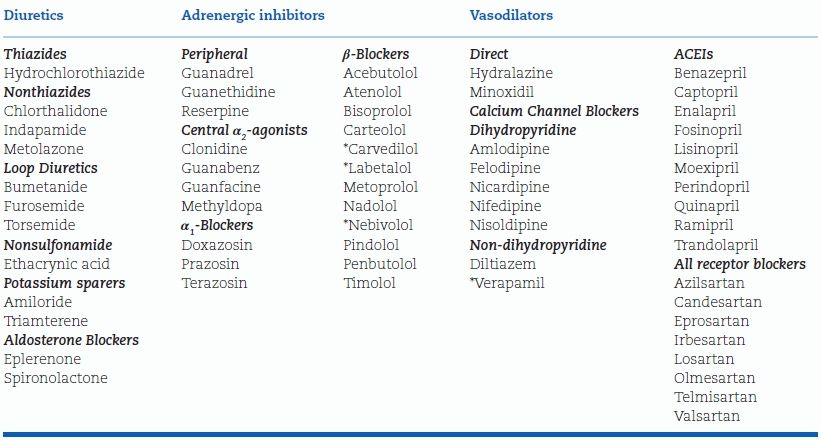
*Vasodilating
There are 10 ACEIs listed, most of them offering no advantage over the others except for longer durations of action than captopril and enalapril. Why so many of the same classes? The answer is given by Light and Lexchin (2012). They state: “Although the pharmaceutical industry and its analysts measure innovation in terms of new molecular entities as a stand-in for therapeutically superior medications, most have provided only minor clinical advantages over existing treatments …. The industry’s own report on all internationally marketed new drugs concluded that only 11% were therapeutically and pharmacologically innovative. Since the mid-1990s, independent reviews have also concluded that about 85% to 90% of all new drugs provide few or no clinical advantages for patients.”
To be sure, in a capitalistic, competitive market system, multiple options within the same classes of drugs will remain available. However, better advice can be provided. Attempts are being made; the National Institute for Clinical Excellence (NICE) in the United Kingdom, the Hypertension Education Program in Canada, the National Institutes of Health (NIH) and, in particular, the National High Blood Pressure Education Program in the U.S. are trying to bring the best advice to practitioners.
The number of hypertensive subjects taking antihypertensive drugs has progressively risen over the past 40 years. Diuretics have continued to be the most commonly prescribed, followed by ACEIs, β-blockers, and CCBs, with ARBs rising the fastest and α-blockers continuing to fall. Logical combinations will be used increasingly (Parati et al., 2014).
Law et al. (2009) suggest that two or three drugs in half usual doses be given, rather than full doses of one or two, both to achieve greater efficacy and to reduce dose-dependent side effects. A “Polypill” containing small doses of a diuretic, ACEI, statin, and aspirin has been shown to be effective (Thom et al., 2013).
The use of drugs in various secondary forms of hypertension (e.g., spironolactone in primary aldosteronism) is considered in the respective chapters on these identifiable causes.
DIURETICS
Among the first orally effective drugs to become available, diuretics are being used even more frequently because their effectiveness has been reiterated and, with lower doses, their side effects minimized.
Diuretics differ in structure and major site of action within the nephron (Fig. 7-1). The site of action determines their relative efficacy, as expressed in the maximal percentage of filtered sodium chloride excreted (Brater, 2000). Agents acting in the proximal tubule (site I) are seldom used to treat hypertension. Treatment is usually initiated with a thiazide-type diuretic (acting at site III, the distal convoluted tubule). Chlorthalidone and indapamide, although they act at the same site, are structurally different from the thiazides and will be covered separately. If renal function is significantly impaired (i.e., serum creatinine exceeding 1.5 mg/dL), a loop diuretic (acting at site II, the thick ascending limb of the loop of Henle) or metolazone likely will be needed. A potassium-sparing agent (acting at site IV) may be given with the diuretic to reduce the likelihood of hypokalemia. By themselves, potassium-sparing agents are relatively weak antihypertensives.
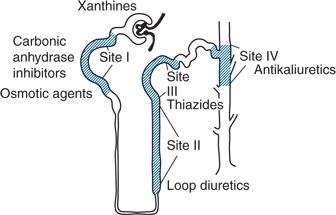
FIGURE 7-1 Diagrammatic representation of the nephron showing the four main tubular sites where diuretics interfere with sodium reabsorption.
The diuretics now available in the U.S. are listed in Table 7-3. Aldosterone receptor blockers, though potassium sparers, are considered separately because of their additional effects.
TABLE 7-3 Diuretics and Potassium-Sparing Agents
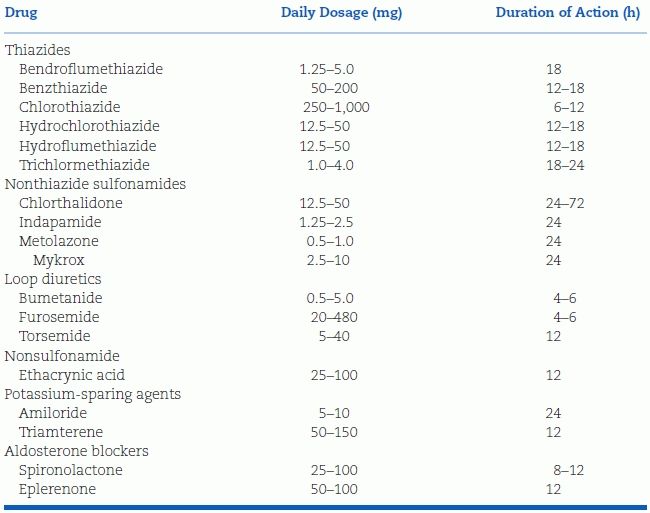
Thiazide Diuretics
Hydrochlorothiazide (HCT) is the most commonly used diuretic for the treatment of hypertension in the U.S. and, if a diuretic is part of a combination product, HCT is almost always chosen (Kaplan, 2011). However, starting in 2004, a group of investigators at the Carver School of Medicine at the University of Iowa have published data showing that chlorthalidone is both stronger and longer-acting than HCT (Carter et al., 2004; Ernst et al., 2011). Their results have been repeatedly confirmed, most impressively with 24-hour ambulatory BP monitoring (Messerli et al., 2011). More importantly, chlorthalidone when used alone has been shown to reduce morbidity and mortality (Roush et al., 2012) whereas HCT when used alone has not been shown to do so (Messerli & Bangalore, 2011). As a consequence of these results, chlorthalidone will likely be used more frequently. One combination of it with an ARB has recently appeared (Cushman et al., 2012a).
Mode of Action
The thiazide diuretics act by inhibiting sodium and chloride cotransport across the luminal membrane of the early segment of the distal convoluted tubule, where 5% to 8% of filtered sodium is normally reabsorbed (Brater, 2000) (see Fig. 7-1, site III). Plasma and extracellular fluid volume are thereby shrunken, and cardiac output falls (Wilson & Freis, 1959). Humoral and intrarenal counterregulatory mechanisms rapidly reestablish the steady state so that sodium intake and excretion are balanced within 3 to 9 days in the presence of a decreased body fluid volume (Sica, 2004a). With chronic use, plasma volume returns partially toward normal, but, at the same time, peripheral resistance decreases (Zhu et al., 2005) (Fig. 7-2).
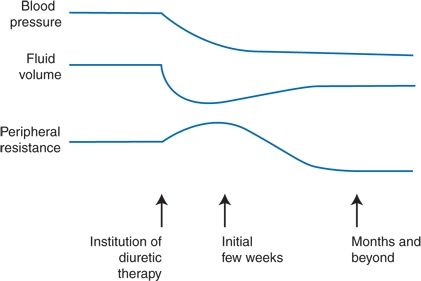
FIGURE 7-2 Scheme of the hemodynamic changes responsible for the antihypertensive effects of diuretic therapy.
Determinants of Response
The degree of BP response to diuretics is predicated on their capacity to activate the counterregulatory defenses to a lower BP and a shrunken fluid volume—in particular, a reactive rise in renin and aldosterone levels. Those who start with low, suppressed plasma renin activity (PRA) and aldosterone levels and who are capable of mounting only a weak rise in these levels after diuretics are initiated have been shown to be more “diuretic responsive” (Chapman et al., 2002). This includes older, black, and hypertensive people, all of whom frequently have lower renin levels (Kaplan, 1977). Those who respond less well, with a fall in mean BP of less than 10%, were found to have a greater degree of plasma volume depletion and greater stimulation of renin and aldosterone, contributing to a persistently high peripheral resistance (van Brummelen et al., 1980). Blockade of the reactive rise in renin–angiotensin–aldosterone, as with the addition of an ACEI or ARB will potentiate the antihypertensive action (McHenry et al., 2013).
Nonthiazide Sulfonamide Diuretics
Chlorthalidone
Although commonly considered a thiazide, chlorthalidone is of a different chemical structure and milligram for milligram is both stronger and longer-acting than is hydrochlorothiazide (HCT), effective even with reduced renal function (Cirillo et al., 2014). In a meta-analysis, Peterzan et al. (2012) found that the estimated dose predicted to reduce SBP by 10 mm Hg was 8.6 mg of chlorthalidone and 26.4 mg of HCT. As noted, although HCT has become by far the most widely used diuretic to treat hypertension in the U.S., chlorthalidone has been used in all trials sponsored by the NIH, with as much or more protection against heart attacks, heart failure, and strokes as seen with other agents (Roush et al., 2012). On the other hand, there are no data showing such benefits of HCT in the currently recommended lower doses of 12.5 to 25 mg/day (Kaplan, 2011).
Indapamide
Indapamide (Lozol) is a chlorobenzene sulfonamide but has a methylindoline moiety, which may provide additional protective actions beyond its diuretic effect (Chillon & Baumbach, 2004). It is as effective in reducing the BP as are thiazides or CCBs (Emeriau et al., 2001); maintains a 24-hour effect; and, in appropriately low doses of 1.25 mg/day, rarely raises serum lipids but in larger doses, hyponatremia and hypokalemia may occur. With 1.5-mg doses, regression of left ventricular hypertrophy (LVH) was better (Gosse et al., 2000) and reduction in microalbuminuria equal to (Marre et al., 2004) that seen with enalapril, 20 mg/day. On the background of an ACEI, indapamide provided a 43% reduction in recurrences of stroke (PROGRESS Collaborative Group, 2001), and it was the diuretic used in the HYVET trial (Beckett et al., 2008).
Metolazone
Metolazone, a long-acting and more potent quinazoline thiazide derivative, maintains its effect in the presence of renal insufficiency (Paton & Kane, 1977). Small doses, 0.5 to 1.0 mg/day, of a new formulation (Mykrox) may be equal to ordinary long-acting thiazide diuretics (Miller et al., 1988); the agent is particularly useful in patients with renal insufficiency and resistant hypertension, but variable absorption may interfere with its efficacy.
Antihypertensive Efficacy
When used alone, diuretics provide efficacy similar to that of other classes of drugs (Law et al., 2009). Blacks and the elderly respond better to diuretics than do nonblacks and younger patients (Brown et al., 2003) presumably because they have lesser renin responsiveness.
Diuretics potentiate the effect of all other antihypertensive agents, including CCBs (Sica, 2004a). This potentiation depends on the contraction of fluid volume by the diuretic (Finnerty et al., 1970a,b) and thereby the prevention of fluid accumulation that frequently follows the use of nondiuretic antihypertensive drugs. Because of the altered pressure–natriuresis curve of primary hypertension (Saito & Kimura, 1996), whenever the BP is lowered, fluid retention is expected (Fig. 7-3). The need for a diuretic may be lessened with ACEIs, ARBs, and DRIs, which inhibit the renin–aldosterone mechanism, and with CCBs, which have some intrinsic natriuretic activity but potentiation persists with all classes.
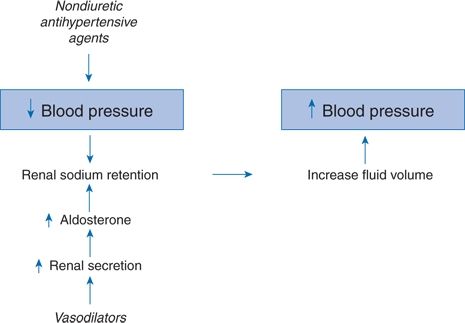
FIGURE 7-3 Manner by which nondiuretic antihypertensive agents may lose their effectiveness by reactive renal sodium retention.
Duration of Action
The durations of action listed in Table 7-3 relate to the diuretic effect; the full antihypertensive effect may not last beyond the diuretic effect. Even though HCT has only a 12- to 18-hour duration of diuretic action, not until the comparison between HCT and chlorthalidone using ambulatory BP monitoring (ABPM) was there a clear evidence for a lessening of HCT antihypertensive effect during the night (Ernst et al., 2006).
Dosage
Monotherapy
The recommended daily dose of thiazide diuretics has been progressively falling from as high as 200 mg of HCT or equivalent doses of other thiazides in the early 1960s (Cranston et al., 1963) to as little as 12.5 mg today. In hypertensives with good renal function, most of the antihypertensive effect will be obtained from such small doses, with less hypokalemia and other side effects. However, as shown by Carlsen et al. (1990), the full antihypertensive effect of low doses of diuretic may not become apparent in 4 weeks, so patience is advised when low doses are prescribed.
Combination Therapy
Thiazides may also be coupled with loop diuretics in those with renal impairment, because they counter the distal nephron hypertrophy that occurs with loop diuretics alone (Brater, 2000). The combination of a thiazide with a loop diuretic usually increases sodium excretion but may induce hypokalemia, hyponatremia, and hypotension (Dussol et al., 2012).
Resistance to Diuretics
Resistance to the natriuretic and antihypertensive action of diuretics may occur for numerous reasons including these:
- Excessive dietary sodium intake.
- For those with renal impairment (i.e., serum creatinine >1.5 mg/dL or creatinine clearance <30 mL/minute), thiazides likely will not work. Because these drugs must be secreted into the renal tubules to work and because endogenous organic acids that build up in renal insufficiency compete with diuretics for transport into the proximal tubule, the renal response progressively falls with increasing renal damage.
- Food affects the absorption and bioavailability of different diuretics to variable degrees (Neuvonen & Kivistö, 1989), so the drugs should be taken in a uniform pattern in terms of the time of day and food ingestion.
- NSAIDs may blunt the effect of most diuretics (Cheng & Harris, 2004).
Protection Against Cardiovascular Events
Diuretics protect against cardiovascular morbidity and mortality as well as any other class of drug (Cushman et al., 2012b). In a network meta-analysis of RCTs published from 1997 through 2009, diuretics were found to be the most effective class of antihypertensive drugs to prevent heart failure (Sciarretta et al., 2011).
Side Effects
As shown in Figure 7-4, the likely pathogenesis for most of the more common complications related to diuretic use arises from the intrinsic activity of the drugs, and most complications are, therefore, related to the dose and duration of diuretic use. Logically, side effects occur with about the same frequency and severity with equipotent doses of all diuretics, and their occurrence will diminish with lower doses.
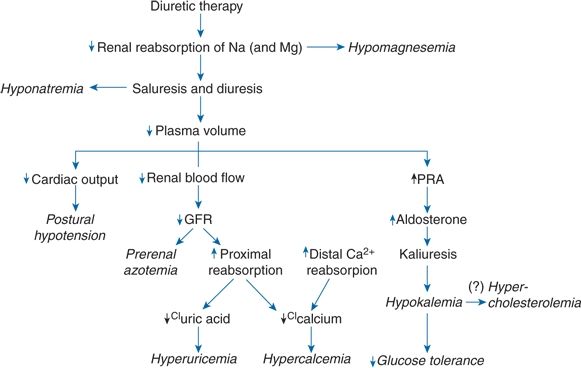
FIGURE 7-4 Mechanisms by which chronic diuretic therapy may lead to various complications. The mechanism for hypercholesterolemia remains in question, although it is shown as arising via hypokalemia. Ca, calcium; CI, chlorine; GFR, glomerular filtration rate; Na, sodium; Mg, magnesium; PRA, plasma renin activity.
Hypokalemia
The degree of hypokalemia is dose dependent. In a meta-analysis that included 26 trials with HCT, 3 of chlorthalidone, and 1 with bendroflumethiazide, Peterzan et al. (2012) showed that the extent of the fall in serum potassium increased with the dose of each drug (Fig. 7-5). The estimated doses to lower serum potassium by 0.4 mmol/L were 11.9 mg for chlorthalidone and 40.5 mg for HCT.
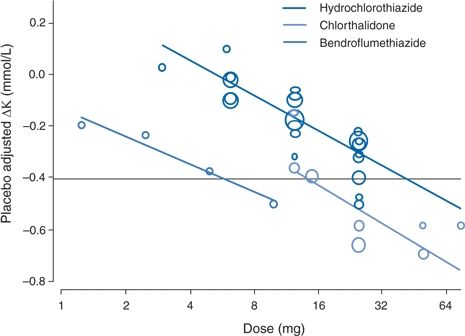
FIGURE 7-5 Dose–response relations between doses of three diuretics and changes in serum potassium. (Adapted from Peterzan MA, Hardy R, Chaturvedi N, et al. Meta-analysis of dose–response relationships for hydrochlorothiazide, chlorthalidone, and bendroflumethiazide on blood pressure, serum potassium, and urate. Hypertension 2012;59:1104–1109.)
The major potential risks of potassium depletion are to increase the incidence of stroke (Levine & Coull, 2002) and ventricular arrhythmias causing sudden death (Grobbee & Hoes, 1995). Patients on digitalis may develop toxicity, perhaps because both digitalis and hypokalemia inhibit the Na+/K+-adenosine triphosphatase (Na+/K+-ATPase) pump, the activity of which is essential to normal intracellular electrolyte balance and membrane potential (Nørgaard & Kjeldsen, 1991).
Ventricular Arrhythmias and Sudden Death
In two case–control studies, the risk of sudden death was nearly doubled in those on large doses of naked diuretics as compared to those on thiazide plus a potassium-sparing agent (Hoes et al., 1995; Siscovick et al., 1994). In the SHEP trial, among those randomly allocated to 12.5 to 25 mg chlorthalidone, the 7.2% who developed hypokalemia had less than half the reduction in major cardiovascular events than did those who remained normokalemic (Franse et al., 2000).
Prevention of Diuretic-Induced Hypokalemia
By lowering dietary sodium, increasing dietary potassium, and using the least amount of diuretic needed, potassium depletion may be avoided. A potassium-sparing agent, β-blocker, ACEI, ARB, or DRI given with the diuretic will reduce the degree of potassium loss but may not prevent the development of hypokalemia. Aldosterone receptor blockers may be more efficient (Coca et al., 2005).
Repletion of Diuretic-Induced Hypokalemia
If prevention does not work, the potassium deficiency can be replaced with supplemental K+, usually given as the chloride. However, potassium citrate (Sakhaee et al., 1991) or bicarbonate (Frassetto et al., 2000) will be more effective in reducing urinary calcium loss in patients with renal stones or osteoporosis. The KC1 may be given as a potassium-containing salt substitute; a number of these substitutes are available, and they are less expensive than are potassium supplements.
Caution is advised in giving potassium supplements to patients receiving ACEIs, ARBs, or DRIs since these will suppress aldosterone levels and thereby prevent the excretion of extra potassium. The problem may be compounded in diabetics who may be unable to move potassium rapidly into cells and in those with renal insufficiency who may have a limited ability to excrete potassium.
Hypomagnesemia
Some of the problems attributed to hypokalemia may be caused by hypomagnesemia but conventional doses of diuretics rarely induce magnesium deficiency (Wilcox, 1999).
Clinical features include weakness, nausea, neuromuscular irritability, and the appearance of ventricular arrhythmias, which are resistant to treatment unless both hypomagnesemia and hypokalemia are corrected (Whang et al., 1985). If repletion is needed, oral magnesium oxide, 200 to 400 mg/day (10 to 20 mmol), or potassium–magnesium citrate may be tolerated without gastrointestinal distress (Pak, 2000).
Hyponatremia
By impairing the dilution of the tubular fluid, thiazides reduce the capacity for rapid and effective elimination of free water, and slight, asymptomatic falls in serum sodium concentration are common (Leung et al., 2011). Rarely, severe, symptomatic hyponatremia develops, usually soon after high doses of diuretics are started in thin elderly women who appear to have an expanded fluid volume from increased water intake in the face of a decreased ability to excrete free water (Mann, 2008).
Hyperuricemia
Serum uric acid levels are high in as many as 30% of untreated hypertensives and diuretics increase renal urate reabsorption, raising uric acid levels further, occasionally provoking gout (Choi et al., 2012). Moreover, Richard Johnson and others have provided evidence for a casual role of hyperuricemia in the pathogenesis of hypertension (Feig et al., 2008a) and renal damage (Obermayr et al., 2008).
Beyond diuretics, Choi et al. (2012) found an association between gout and the use of β-blockers, ACEIs, and ARBs except losartan, whereas the use of losartan or CCBs was associated with a lower risk. If therapy is given for hyperuricemia, the logical choice is probenecid to increase renal excretion of uric acid and possibly to lower the BP (Soletsky & Feig, 2012).
Calcium Metabolism Alterations
Renal calcium reabsorption also is increased with chronic thiazide therapy, and urinary calcium excretion is decreased by 40% to 50% (Friedman & Bushinsky, 1999). A slight rise in serum calcium (i.e., 0.1 to 0.2 mg/dL) is usual, and hypercalcemia is often provoked in patients with preexisting hyperparathyroidism or vitamin D–treated hypoparathyroidism. By reducing renal calcium excretion, thiazides are used to treat patients with renal stones caused by hypercalcemia from increased calcium absorption (Quereda et al., 1996). The retention of calcium in bone offers protection from osteoporosis and fractures (Schoofs et al., 2003). However, loop diuretics, which increase urinary calcium excretion, are associated with an increased rate of hip bone loss in older men (Lim et al., 2008).
Glucose Intolerance and Insulin Resistance
Insulin resistance, impairment of glucose tolerance, precipitation of overt diabetes, and worsening of diabetic control have all been observed in patients taking larger doses of thiazides (Carter et al., 2008b). In a review of data from 83 trials with thiazides, the rise in blood glucose was closely correlated with the fall in serum potassium (Zillich et al., 2006).
As with all the adverse effects of diuretics, the impairment of glucose utilization that connotes insulin resistance is seen more with high doses (McHenry et al., 2013). The incidence of new-onset diabetes among the ALLHAT trial participants who took chlorthalidone (most at a dose of 25 mg) was 11.5% compared to 8.3% in those who started with amlodipine and 7.6% in those who started with lisinopril (Black et al., 2008). It is likely that part of the increases in diabetes in diuretic-treated patients comes from the concomitant use of β-blockers, the “conventional” therapy of older trials.
Effect on Lipids
With low doses, thiazides have little effect on the blood lipid profile (Weir & Moser, 2000). However, higher doses may induce significant effects on fat distribution, which in turn may be associated with insulin resistance. Eriksson et al. (2008) examined the effects of a placebo, the ARB candesartan (16 to 32 mg/day) and HCT (50 mg/day), each given to 26 hypertensives with abdominal obesity for 12 weeks in a randomized crossover design. After the 12 weeks on the diuretic, the subjects had increases in abdominal and hepatic fat, abnormal liver function test, insulin resistance, and increased C-reactive protein levels. None of these effects were seen after placebo or ARB.
Erectile Dysfunction
Impotence may be more common with diuretics than with other drugs. In the large, randomized Medical Research Council (MRC) trial, impotence was reported by 22.6% of the men on bendrofluazide, as compared to a rate of 10.1% among those on placebo and 13.2% among those on propranolol (Medical Research Council Working Party, 1981). In the Treatment of Mild Hypertension Study (TOMHS), the men randomized to chlorthalidone had a 17.1% incidence of erection problems through 24 months, as compared to an 8.1% incidence in those on placebo (Grimm et al., 1997).
Other Side Effects
Fever and chills, blood dyscrasias, cholecystitis, pancreatitis, necrotizing vasculitis, acute interstitial nephritis, and noncardiogenic pulmonary edema have been seen rarely. Allergic skin rashes occur in 0.28% of patients, and approximately the same percentage develops photosensitivity, which may be involved in the reported increase in lip cancer (Friedman et al., 2012). An increased relative risk of renal cell cancer has been reported with diuretic therapy (Corrao et al., 2007), but the association is much stronger with hypertension per se (Colt et al., 2011).
Conclusion
Strong controlled trial data document the benefits of diuretics in particular chlorthalidone, for the treatment of hypertension. Nonetheless, diuretics can cause multiple metabolic perturbations that could reduce their ability to protect against progressive atherosclerosis as they lower BP, including rises in uric acid, increasing insulin resistance, and deranged fat distribution. These adverse effects are dose dependent and should be much less problematic with appropriately lower doses, doses that will provide most, if not all, of their antihypertensive effects.
Loop Diuretics
Loop diuretics primarily block chloride reabsorption by inhibition of the Na+/K+/Cl− cotransport system of the luminal membrane of the thick ascending limb of Henle loop, the site where 35% to 45% of filtered sodium is reabsorbed (see Fig. 7-1). Therefore, the loop diuretics are more potent and have a more rapid onset of action than do the thiazides. However, they are no more effective in lowering BP or less likely to cause side effects if given in equipotent amounts. Their major use is in patients with renal insufficiency, in whom large enough doses can be given to achieve an effective luminal concentration (see Chapter 9).
Furosemide
The maintenance of a slightly shrunken body fluid volume, which is critical for an antihypertensive action from diuretic therapy, is not met by the short duration of furosemide action (3 to 6 hours for an oral dose); during the remaining hours, sodium is retained, so that net fluid balance over 24 hours is left unaltered (Wilcox et al., 1983). If furosemide is used twice daily, the first dose should be given early in the morning and the second in the late afternoon, both to provide diuretic action at the time of sodium intake and to avoid nocturia.
Bumetanide
Bumetanide, although 40 times more potent and 2 times more bioavailable than furosemide on a weight basis, is identical in its actions when given in an equivalent dose (Brater et al., 1983).
Torsemide
Torsemide differs from the other diuretics in that it is mainly eliminated by hepatic metabolism, with only 20% being excreted unchanged in the urine (Brater, 1993). Therefore, it has a more prolonged duration of action, as long as 12 hours.
In small doses of 2.5 to 5 mg, torsemide may lower BP in uncomplicated hypertension, whereas larger doses are needed for chronic edematous states or with renal insufficiency (Dunn et al., 1995). In patients with chronic renal disease, 40 mg of torsemide once a day provided equal natriuresis and hypertensive effect as 40 mg of furosemide twice a day (Vasavada et al., 2003).
Ethacrynic Acid
Although structurally different from furosemide, ethacrynic acid also works primarily in the ascending limb of Henle loop and has an equal potency. It is used much less than is furosemide, mainly because of its greater propensity to cause permanent hearing loss with high doses. Since it does not contain a sulfonamide moiety, its main use has been in patients with sulfonamide sensitivity.
Potassium-Sparing Agents
Amiloride and triamterene act directly to inhibit sodium reabsorption by the epithelial sodium channels in the renal distal tubule, decreasing the net negative potential in the tubular lumen and thereby reducing potassium and hydrogen secretion and excretion, independent of aldosterone. Since neither are potent natriuretics, they are almost exclusively used in combination with thiazides, which, by delivering more sodium to the K+ sparers’ site of action, increase their K+-sparing effect while countering the K+-wasting effect of the diuretic. Presumably, by preventing hypokalemia, the use of K+-sparing diuretics reduced the risk of death compared to the use of non–K+-sparing diuretics (Hebert et al., 2008).
Amiloride
Amiloride is usually used with a thiazide diuretic in tablets containing 50 mg of HCT and 5 mg of amiloride. The drug has been used as medical therapy for hyperaldosteronism in patients intolerant to aldosterone blockers and in patients with mutations of the genes regulating sodium channels that lead to the full-blown Liddle syndrome (see Chapter 11) or to a less severe prototype from the T594M polymorphism (Baker et al., 2002).
Nausea, flatulence, and skin rash have been the most frequent side effects and hyperkalemia the most serious. Moreover, a number of cases of hyponatremia in elderly patients have been reported after its use in combination with HCT (Mathew et al., 1990).
Triamterene
As with amiloride, triamterine (37.5 mg) is usually combined with HCT (25 mg). Triamterene may be excreted into the urine and may find its way into renal stones (Sörgel et al., 1985). Because triamterene is a folic acid antagonist, it should not be used during pregnancy (Hernández-Díaz et al., 2000).
Aldosterone Receptor Blockers
The first of these, spironolactone, has long been available but little used in the U.S. until publication of the Randomized Aldactone Evaluation Study (RALES) which showed a 30% decrease in mortality in patients with severe heart failure given 25 mg of spironolactone in addition to their other medications (Pitt et al., 1999). Since then, a large body of experimental and clinical evidence has revealed a multiorgan profibrotic effect of aldosterone so that blocking the hormone has assumed an important place in clinical medicine. At the same time, the marketing of a more specific aldosterone blocker, eplerenone, has stimulated the use of these agents.
Mode of Action
The primary mineralocorticoid aldosterone causes hypertension when present in large excess, the syndrome of primary aldosteronism covered in Chapter 11. However, even “normal” amounts of aldosterone in the presence of the relatively high sodium intake of modern societies are now known to activate mineralocorticoid receptors in multiple organs including the brain, heart, kidney, and blood vessels (Schiffrin, 2006). In turn, vasculitis and fibrosis are induced, independent of the traditional renal sodium–retaining effect of the hormone. Moreover, the incidence of hypertension over 4 years was 60% higher in those initially nonhypertensive subjects who were in the highest quartile of serum aldosterone (Vasan et al., 2004).
Eplerenone in a twice higher dose has equivalence to spironolactone in blocking the mineralocorticoid receptor but a much lower blockade of androgen and progesterone receptors, explaining its fewer side effects (Funder, 2002). In 2003, the addition of eplerenone was shown to reduce morbidity and mortality among patients with acute myocardial infarction (MI) complicated by left ventricular (LV) dysfunction in the Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS) (Pitt et al., 2003). Subsequently, the drug was shown to reduce mortality in these subjects whether they had been hypertensive or not (Pitt et al., 2008).
In both the RALES and EPHESUS trials, the aldosterone blocker provided additional benefit to patients receiving full doses of blockers of the renin–angiotensin system (RAS), ACEIs, or ARBs. It is now known that aldosterone synthesis is not completely suppressed with these agents, breaking through to maintain the pretreatment aldosterone levels even if angiotensin II (AII) levels remain suppressed (Bomback et al., 2012).
Antihypertensive Efficacy
Spironolactone has been used alone to treat hypertension for many years, particularly in France (Jeunemaitre et al., 1987), but its major use in the U.S. has been as a K+ sparer in combination with a thiazide diuretic, providing an effect equivalent to 32 mmol of KC1 (Toner et al., 1991) or to treat aldosteronism caused by bilateral adrenal hyperplasia. More recently, it has been found to effectively control patients with refractory hypertension (Chapman et al., 2007; Oxlund et al., 2013). As expected, the drug lowers BP more in patients with low plasma renin and higher aldosterone levels (Weinberger, 2004). Aldosterone blockers improve diastolic function (Grandi et al., 2002), are antiarrhythmic (Swedberg et al., 2012), reduce proteinuria in patients with diabetic nephropathy (Sato et al., 2003), and prevent diuretic-induced sympathetic nervous system activation and insulin resistance (Raheja et al., 2012). For these reasons, the use of aldosterone blockers will almost certainly expand to initial therapy, usually in combination with a diuretic, for more and more hypertensives.
Side Effects
The less specific spironolactone in doses of 25 to 50 mg/day induced gynecomastia in 6% of patients and biochemical abnormalities (mainly hyperkalemia) in 2% of the patients with resistant hypertension in the ASCOT trial (Chapman et al., 2007). The more specific eplerenone induced gynecomastia in fewer than 1% of men in the EPHESUS trial (Pitt et al., 2005). Hyperkalemia may occur with either agent but is more common in the presence of renal insufficiency; concomitant β-blocker, ACEI, ARB, or DRI therapy; or the use of potassium supplements (Muzzarelli et al., 2012). With appropriate monitoring, eplerenone is both safe and effective in patients with impaired renal function (Eschalier et al., 2013). The antibiotic trimethoprim is similar to the potassium-sparing agent amiloride and reduces potassium excretion by 40%. Therefore, hyperkalemia may occur if the antibiotic is given to patients on an aldosterone blocker (Antoniou et al., 2011).
ADRENERGIC-INHIBITING DRUGS
Of the adrenergic-inhibiting agents currently used to treat hypertension, some act centrally on α2-receptors to inhibit sympathetic nerve activity, some inhibit postganglionic sympathetic neurons, and some block the α- or β-adrenoreceptors on target organs (Fig. 7-6). Agents that act by blocking ganglia are no longer used.
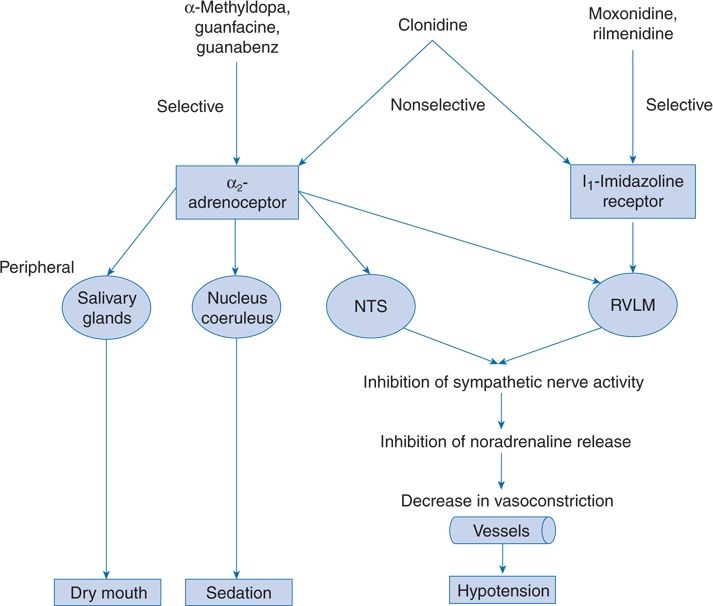
FIGURE 7-6 Central antihypertensive mechanisms of various types of centrally acting antihypertensive drugs. NTS, nucleus tractus solitarii; RVLM, rostral ventrolateral medulla. (Modified from van Zwieten PA. The renaissance of centrally acting antihypertensive drugs. J Hyperten 1999;17(Suppl 3):S15–S21.)
Central α-Agonists
Central α-agents stimulate α2a-adrenergic receptors that are involved in depressor sympathoinhibitory mechanisms (Vongpatanasin et al., 2011). Some are selective, whereas clonidine also acts on central imidazoline receptors. These drugs have well-defined effects, including
- A marked decline in sympathetic activity reflected in lower levels of norepinephrine (NE)
- A reduction of the ability of the baroreceptor reflex to compensate for a decrease in BP, accounting for the relative bradycardia and enhanced hypotensive action noted on standing
- A modest decrease in both peripheral resistance and cardiac output
- A fall in plasma renin levels
- Fluid retention
- Maintenance of renal blood flow despite a fall in BP
- Common side effects reflecting their central site of action: Sedation, decreased alertness, and a dry mouth
Methyldopa
From the early 1960s to the late 1970s, when β-blockers became available, methyldopa was the second most popular drug (after diuretics) used to treat hypertension. Now its use is almost exclusively for the treatment of hypertension during pregnancy.
Methyldopa is the α-methylated derivative of dopa, the natural precursor of dopamine and NE. Its mode of action involves the formation of methylnorepinephrine, which acts as a potent agonist at α-adrenergic receptors within the central nervous system (CNS) (van Zwieten, 1999).
Antihypertensive Efficacy
BP is lowered maximally approximately 4 hours after an oral dose of methyldopa, and some effect persists for up to 24 hours. For most patients, therapy should be started with 250 mg two times per day, and the daily dosage can be increased to a maximum of 3.0 g on a twice-per-day schedule. In patients with renal insufficiency, the dosage should be halved.
Side Effects
A variety of autoimmune side effects, including fever and liver dysfunction, can occur with methyldopa. Liver dysfunction usually disappears when the drug is stopped, but at least 83 cases of serious hepatotoxicity were reported by 1975 (Rodman et al., 1976), with diffuse parenchymal injury similar to autoimmune chronic active hepatitis (Lee, 1995).
An impairment of psychometric performance (Johnson et al., 1990) and a selective loss of upper airway motor activity (Lahive et al., 1988) may not be obvious until the drug is stopped. Overall, in large surveys, the number and range of the adverse reactions to methyldopa are impressive (Webster & Koch, 1996). In view of its unique and potentially serious side effects, other central α-agonists should be used in place of methyldopa.
Guanabenz
Guanabenz, an aminoguanidine, works like clonidine and methyldopa and causes similar side effects. Therapy should begin with 4 mg twice per day, with increments up to a total of 64 mg/day.
Guanfacine
Another selective central α2-agonist, guanfacine appears to enter the brain more slowly and to maintain its antihypertensive effect longer than guanabenz, translating into a once-per-day dosage and perhaps fewer CNS side effects (Lewin et al., 1990). Withdrawal symptoms are less common than with clonidine. These characteristics make it the most attractive of this group of centrally acting α2-agonists.
Clonidine
Clonidine acts centrally on both α2-receptors and imidazoline receptors (see Fig. 7-6). When taken orally, the BP begins to fall within 30 minutes, with the greatest effect occurring between 2 and 4 hours. The duration of effect is from 8 to 12 hours, so it should be given three times a day. The starting dose may be as little as 0.075 mg (Clobass Study Group, 1990), with a maximum of 1.2 mg/day.
A transdermal preparation that delivers clonidine continuously over a 7-day interval is effective and causes milder side effects than does oral therapy (Giugliano et al., 1998), but it may cause considerable skin irritation and side effects similar to those seen with the oral drug, including rebound hypertension when discontinued. It is available in doses of 0.1, 0.2, and 0.3 mg/day.
Side Effects
Clonidine shares the two most common side effects, sedation and dry mouth with methyldopa but not the autoimmune hepatic and hematologic derangements. Depression of sinus and atrioventricular (AV) nodal function may be common, and a few cases of severe bradycardia have been reported (Byrd et al., 1988).
Rebound and Discontinuation Syndromes
If any antihypertensive therapy is inadvertently stopped abruptly, various discontinuation syndromes may occur: (a) A rapid asymptomatic return of the BP to pretreatment levels, which occurs in the majority of patients; (b) a rebound of the BP plus symptoms and signs of sympathetic overactivity; and (c) an overshoot of the BP above pretreatment levels.
A discontinuation syndrome has been reported, more frequently with clonidine (Neusy & Lowenstein, 1989), likely reflecting a rapid return of catecholamine secretion that had been suppressed during therapy. Those who had been on a combination of a central adrenergic inhibitor, e.g., clonidine, and a β-blocker may be particularly susceptible if the central inhibitor is withdrawn while the β-blocker is continued (Lilja et al., 1982). This leads to a sudden surge in plasma catecholamines in a situation in which peripheral α-receptors are left unopposed to induce vasoconstriction because the β-receptors are blocked and cannot mediate vasodilation.
If a discontinuation syndrome appears, clonidine should be restarted, and the symptoms will likely recede rapidly.
Other Uses
Clonidine has been reported to be useful in numerous conditions that may accompany hypertension, including
- Restless legs syndrome (Wagner et al., 1996)
- Opiate withdrawal (Bond, 1986)
- Menopausal hot flashes (Pandya et al., 2000)
- Diarrhea due to diabetic neuropathy (Fedorak et al., 1985)
- Sympathetic nervous hyperactivity in patients with alcoholic cirrhosis (Esler et al., 1992)
- However, the use of clonidine perioperativel has been found to be dangerous (Devereaux et al., 2014).
Imidazoline Receptor Agonists
Not available in the U.S. but used elsewhere, monoxidine and rilmenidine are two centrally acting drugs that have as their primary site of action the imidazoline receptor located in the rostral ventrolateral medulla oblongata, wherein α2-receptors are less abundant (see Fig. 7-6) (van Zwieten, 1999). They effectively reduce sympathetic activity (Esler et al., 2004) with less of the sedation and dry mouth seen with clonidine and selective α2-agonists.
Peripheral Adrenergic Inhibitors
Reserpine
First reported to be an effective antihypertensive in the 1940s (Bhatia, 1942), reserpine became a popular drug in the 1960s and 1970s but has been used less and less because, being an inexpensive generic, it has no constituency pushing for its use, and when used in high doses, it has caused depression, earning it a bad reputation.
Reserpine, one of the many alkaloids of the Indian snakeroot Rauwolfia serpentina, is absorbed readily from the gut, is taken up rapidly by lipid-containing tissue, and binds to sites involved with storage of biogenic amines. Its effects start slowly and persist, so only one dose per day is needed.
Reserpine blocks the transport of NE into its storage granules so that less of the neurotransmitter is available when the adrenergic nerves are stimulated, resulting in a decrease of peripheral vascular resistance. Catecholamines also are depleted in the brain, which may account for the sedative and depressant effects of the drug, and in the myocardium, which may decrease cardiac output and induce a slight bradycardia.
Antihypertensive Efficacy
By itself, reserpine has limited antihypertensive potency, resulting in an average decrease of only 3/5 mm Hg; when combined with a thiazide, the reduction averaged 14/11 mm Hg (Veterans Administration Cooperative Study, 1962). With a diuretic, as little as 0.05 mg once daily will provide most of the antihypertensive effect of 0.25 mg and is associated with less lethargy and impotence (Participating VA Medical Centers, 1982).
Side Effects
Side effects, which are relatively infrequent at appropriately low doses include nasal stuffiness, increased gastric acid secretion, and CNS depression, which may simply tranquilize an apprehensive patient and is rarely severe enough to lead to serious depression.
Guanethidine
Guanethidine at one time was frequently used because it requires only one dose per day and has a steep dose–response relationship, thus producing an effect in almost every patient. Because of severe postural hypotension, the use of guanethidine has virtually disappeared.
α-Adrenergic Receptor Blockers
Selective α1-blockers have had a relatively small share of the overall market for antihypertensive drugs in the U.S., and as a consequence of the increase in heart failure reported in the ALLHAT trial (ALLHAT Officers, 2000), their use in the U.S. is now almost exclusively for relief of prostatism. Nonetheless, doxazosin was successfully used in the ASCOT trial without an increase in heart failure (Chapman et al., 2008).
Mode of Action
The nonselective α-blockers phenoxybenzamine and phentolamine are used almost exclusively in the medical management of pheochromocytoma, because they are only minimally effective in primary hypertension (see Chapter 12).
Prazosin, doxazosin, and terazosin act as a competitive antagonist of postsynaptic α2-receptors (Fig. 7-7). These agents block the activation of postsynaptic α1-receptors by circulating or neurally released catecholamines, reducing peripheral resistance without major changes in cardiac output.
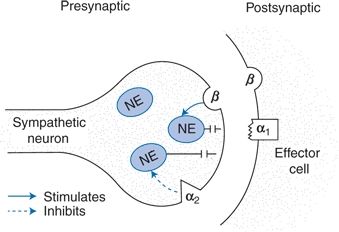
FIGURE 7-7 Schematic view of the action of selective postsynaptic α1-blockers. By blocking the α1-adrenergic receptor on the vascular smooth muscle, catecholamine-induced vasoconstriction is inhibited. The α2-adrenergic receptor on the neuronal membrane is not blocked; therefore, inhibition of additional NE release by the short feedback mechanism is maintained.
The presynaptic α2-receptors remain open, capable of binding neurotransmitter and thereby inhibiting the release of additional NE through a direct negative-feedback mechanism. This inhibition of NE release explains the lesser frequency of tachycardia, increased cardiac output, and rise in renin levels that characterize the response to drugs that block both the presynaptic α2-receptor and the postsynaptic α1-receptor (e.g., phentolamine). Despite this selective blockade, neurally mediated responses to stress and exercise are unaffected, and the baroreceptor reflex remains active.
Accompanying these desirable attributes may be other actions that lessen the usefulness of α-adrenergic blockers: They relax the venous bed as well and, at least initially, may affect the visceral vascular bed more than the peripheral vascular bed. The subsequent pooling of blood in the viscera may explain the propensity to first-dose hypotension seen with the fast-acting prazosin (Saxena & Bolt, 1986). Volume retention is common, perhaps because renin and aldosterone levels are less suppressed than they are with other adrenergic-inhibiting drugs.
Prazosin is rapidly absorbed, reaches maximal blood levels at 2 hours, and has a plasma half-life of approximately 3 hours. Terazosin and doxazosin are less lipid soluble and have half, or less, of the affinity for α1-receptors as compared with prazosin. Therefore, they induce a slower and less profound initial fall in BP, particularly after standing, than does prazosin.
Antihypertensive Efficacy
The antihypertensive efficacy of doxazosin and terazosin is equivalent to that of diuretics, β-blockers, ACEIs, and CCBs (Achari et al., 2000). The addition of doxazosin was shown in the ASCOT trial to control hypertension effectively in patients resistant to two or more other agents (Chapman et al., 2008).
The initial dose should be 1 mg, slowly titrated upward to achieve the desired fall in BP, with a total daily dose of up to 20 mg. α-Blockers can be given at bedtime to provide a greater nocturnal fall in BP in and blunting of the morning surge that is involved in the increased incidence of cardiovascular events at that time (Matsui et al., 2008).
Genitourinary Function
Doxazosin, tamsulosin, and terazosin have been found to provide excellent relief from the obstructive symptoms of benign prostatic hypertrophy. The combination of doxazosin and the 5α-reductase inhibitor finasteride slowed the clinical progression of benign prostatic hypertrophy (BPH) better than either drug alone (McConnell et al., 2003).
In the TOMHS trial of a representative from each of the five major classes of antihypertensives, only doxazosin reduced the incidence of impotence below that seen with placebo (Grimm et al., 1997).
The ALLHAT experience indicates the need to use a diuretic with an α-blocker for the treatment of hypertension, particularly in those with LVH or other risk factors for congestive heart failure (CHF) (Matsui et al., 2008). α-Blockers are useful as add-on therapy in patients with resistant hypertension and the preferred initial therapy for hypertensives with BPH.
Side Effects
Postural hypotension developing in 30 to 90 minutes may be seen particularly in volume-depleted patients given the shorter-acting prazosin. Urinary incontinence in women may be caused by α-blockers (Marshall & Beevers, 1996).
β-Adrenergic Receptor Blockers
For many years, β-adrenergic blocking agents were the second most popular antihypertensive drugs after diuretics. Although they are no more effective than other antihypertensive agents and may on occasion induce serious side effects, they offer the special advantage of relieving a number of concomitant diseases. In view of their proven ability to provide secondary cardioprotection after an acute MI, it was hoped that they would provide special primary protection against initial coronary events as well. This hope remains unfulfilled. To the contrary, β-blockers have failed to reduce heart attacks better than other classes (Bangalore et al., 2012) while providing less protection against stokes (Law et al., 2009). This is particularly true for the most popular, atenolol (Lindholm et al., 2005).
Nonetheless, the proven benefits of β-blockers in patients with either coronary disease (particularly after an acute MI) or CHF ensure that these drugs will continue to be widely used. Moreover, the use of the vasodilating β-blockers, covered after the traditional ones, will continue to replace the traditional ones.
Mode of Action
These agents are chemically similar to β-agonists and to each other (Fig. 7-8). The competitive inhibition of β-blockers on β-adrenergic receptors produces numerous effects on functions that regulate the BP, including a reduction in cardiac output, a diminution of renin release, perhaps a decrease in central sympathetic nervous outflow, and a presynaptic blockade that inhibits catecholamine release. The hemodynamic effects appear to change over time. Cardiac output usually falls acutely (except with high-ISA [intrinsic sympathomimetic activity] pindolol) and remains lower chronically; peripheral resistance, on the other hand, usually rises acutely but falls toward, if not to, normal with time (Man in’t Veld et al., 1988).
FIGURE 7-8 Structure of propranolol and the β-agonist isoproterenol.
Pharmacologic Differences
Since the introduction of propranolol for treatment of hypertension in 1964 (Prichard and Gillam, 1964), a number of similar drugs have been synthesized, approximately 20 being marketed throughout the world, 12 in the U.S. The various β-blockers can be conveniently classified by their relative selectivity for the β1-receptors (primarily in the heart) and the presence of ISA, also referred to as partial agonist activity and their lipid solubility (Table 7-4).
TABLE 7-4 Pharmacologic Properties of Some β-Blockers
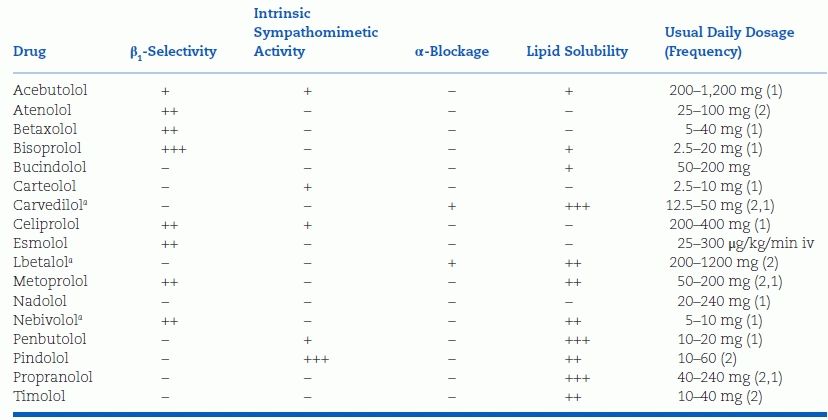
Parenthesis (2,1) indicate short- and long-acting formulations.
+, ++, and +++ signs indicate the magnitude of the effect on various properties; − sign indicates no effect.
aVasodilating.
Lipid Solubility
Those that are more lipid soluble (lipophilic) tend to be taken up and metabolized extensively by the liver. As an example, with oral propranolol and metoprolol, up to 70% is removed on the first pass of portal blood through the liver. The bioavailability of these β-blockers is, therefore, less after oral than after intravenous administration.
Those such as nadolol, which is much less lipid soluble (lipophobic), escape hepatic metabolism and are mainly excreted by the kidneys, unchanged. As a result, its plasma half-life and duration of action is much longer.
β1-Receptor Cardioselectivity
All currently available β-blockers antagonize cardiac β1-receptors competitively, but they vary in their degree of β2-receptor blockade in extracardiac tissues. The assumption that an agent with relative cardioselectivity is automatically less likely to cause side effects must be tempered by these considerations: Recognizing that no β-blocker is purely cardioselective, particularly in large doses, and when high endogenous catechol levels are needed, as during an attack of asthma, even minimal degrees of β2-blockade from a cardioselective drug may cause trouble (Haffner et al., 1992). However, more cardioselective β-blockers have been found to be more protective against strokes than less cardioselective ones (Webb et al., 2011).
On the other hand, in the presence of certain concomitant diseases, such as migraine and tremor, a nonselective β2-antagonist effect may be preferable.
Intrinsic Sympathomimetic Activity
Of the β-blockers now available in the U.S., pindolol and, to a lesser degree, acebutolol have ISA, implying that even in concentrations that fully occupy the β-receptors, the biologic effect is less than that seen with a full agonist.
Antihypertensive Efficacy
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


