Treatment of Myoclonic Epilepsies in Infancy and Early Childhood
Raman Sankar*
James W. Wheless†
Charlotte Dravet‡
Renzo Guerrini§
Marco T. Medina¶
Michelle Bureau‡
Pierre Genton‡
Antonio V. Delgado–Escueta¶
*Departments of Neurology and Pediatrics, David Geffen School of Medicine at UCLA and Mattel Children’s Hospital at UCLA, Los Angeles, California
†Departments of Neurology and Pediatrics, University of Texas School of Medicine, Houston, Texas
§Epilepsy, Neurophysiology, Neurogenetics Unit, Department of Child Neurology and Psychiatry, University of Pisa and Research Institute ‘Stella Maris’ Foundation, Pisa Italy
¶Comprehensive Epilepsy Program and Epilepsy Center of Excellence at VA GLAHS, David Geffen School of Medicine at UCLA, Los Angeles, California, ||National Autonomous University of Honduras, Tegucigalpa, Honduras
‡Centre Saint-Paul- Hôpital Henri Gastaut, Marseille, France
¶Comprehensive Epilepsy Program and Epilepsy Center of Excellence at VA GLAHS, David Geffen School of Medicine at UCLA, Los Angeles, California
Introduction
As described amply in the many chapters of this book, the myoclonic epilepsies comprise a very broad spectrum of clinical disorders in which myoclonus is an important epileptic manifestation. Their etiologies can range from the so-called idiopathic varieties (many of which are now revealing themselves to be channelopathies, receptoropathies, and modulators/sensors of ion channels) to others with neurodegenerative features— these latter again comprising a heterogeneous group. While this book addresses itself to the idiopathic myoclonic epilepsies, the medications used to treat myoclonic seizures do not distinguish between idiopathic and symptomatic myoclonic syndromes. In traditional thinking, idiopathic epilepsies have been thought of as genetic epilepsies without a structural and metabolic basis, and generally not associated with major neurologic deficits. There are now beginning to be attributed to altered excitability resulting from mutations of ion channels receptors or sensors/modulators of ion channels. An epilepsy that is accompanied by severe, medically refractory seizures and mental retardation was classified as cryptogenic, rather than idiopathic, if no structural or metabolic etiology is identified according to the International League Against Epilepsy. The discovery of different germ line missense mutations of SCNA1A to be associated with GEFS+ and de novo somatic “stop” mutations of SCNA1 in Dravet syndrome certainly challenges our traditional use of the term cryptogenic. Regardless of such semantic diversions, there is a need for screening strategies to facilitate drug discovery for the symptomatic treatment of myoclonic seizures. Ultimately, development of drugs designed against specific genetic mutations or specific epilepsy syndromes will need to be pursued.
Animal models of myoclonic seizures have played a role in evaluating the possible efficacy of drug therapy for treatment of human myoclonic seizures. Animal models can be classified as those involving provoked seizures and those involving genetic models. Provocation of seizures in the former is commonly accomplished by chemical or electrical means, although other means, such as hypoxia and intermittent light stimulation, have also been used. Classic pharmacologic screening involving rodents, as used by the Anticonvulsant Screening Project of the National Institute of Neurologic Disorders and Stroke, has produced inconsistent information for predicting the efficacy of potential therapeutic agents against myoclonus. With earlier-generation antiepileptic drugs (AEDs), the models used were only somewhat predictive of efficacy. Only clonazepam (CZP) and valproic acid (VPA) have demonstrated efficacy in animal models for protection against all three seizure provocations involving subcutaneous (sc) pentylenetetrazole, bicuculline, and picrotoxin (1). Ethosuximide, which was only marginally active against bicuculline, has been shown not to be a useful antimyoclonic AED (1,2). Phenytoin (PHT) and carbamazepine (CBZ) clearly distinguished themselves as agents that were mainly active in the maximal electroshock model (3). Among new-generation AEDs, felbamate has shown clinical utility in a variety of myoclonic seizures and was also active in protecting rats against sc pentylenetetrazole, sc bicuculline, and sc picrotoxin (4). Gabapentin (GBP), the next agent to arrive on the United States market, has been shown to be ineffective in sc bicuculline and picrotoxin provocations in the rodent (5) and appears to be clinically useful only for partial and secondarily generalized seizures (6). Vigabatrin (VGB), like GBP, has not demonstrated significant activity against sc bicuculline or picrotoxin challenge, and is not a clinically useful agent for most syndromes involving myoclonic seizures (7). Experience with GBP highlights another difficulty in the use of animal models for screening compounds to treat myoclonus. Kanthasamy et al. (8) found GBP to be antimyoclonic in a posthypoxic rodent model, but not for myoclonus induced by administration of p,pʹ-DDT [1,1,1-trichloro-2,2-bis(p-chlorophenyl)ethane].
Experience with other new-generation AEDs has also revealed many inconsistencies. Lamotrigine (LTG), which has an animal testing profile resembling that of PHT (9), has demonstrated clinical utility for treating some patients with juvenile myoclonic epilepsy (JME) (10). Paradoxically, LTG is also known to exacerbate myoclonus in some patients (11,12,13). Topiramate (TPM), another new-generation agent, has been shown to be ineffective in blocking chemically induced seizures (14). Nevertheless, this medication may be of value in treating a variety of syndromes that involve myoclonus, as is discussed later in this chapter.
The most extensively described genetic model of epilepsy involves Papio papio, the photosensitive baboon. Killiam et al. (15) first reported that when these animals were subjected to intermittent photic stimulation (IPS), paroxysmal discharges in the form of spikes and waves or polyspikes and waves resulted. Behaviorally, bilateral and synchronous myoclonic jerks appeared and were followed by generalized convulsive seizures. The involvement of the GABAergic system in this model was described in detail by Meldrum and Wilkins (16). This model correctly predicted lack of efficacy of GBP in human myoclonic epilepsies (17). It also predicted efficacy of ethosuximide at high doses in myoclonic epilepsies (18,19). However, vigabatrin (VGB), was shown to be effective in blocking photically induced epileptic activity in the baboon (18), suggesting that the baboon model also has limitations in its ability to predict the clinical utility of test AEDs. This model is apparently not routinely employed in screening and many of the new-generation AEDs have no published literature describing their use in this model.
Another well-characterized genetic model involves the tottering mouse. The tg locus causes a delayed-onset recessive neurologic disorder in the mouse, featuring a stereotyped triad of ataxia, intermittent absence seizures, and bursts of generalized 6- to 7-Hz spike waves. This model has not been routinely used in screening for AEDs; hence, its reliability for this purpose is unknown. Other genetic models of inherited myoclonus include a disorder of the inhibitory glycine receptor that results in spontaneous and stimulus-sensitive myoclonus in Polled Hereford calves (19) and spontaneous and photically induced myoclonus in a mutant strain of Fayoumi chickens (20). The possible importance of the glycine receptor in some myoclonic disorders is highlighted by the recent development of a transgenic mouse model of hypereklexia, which involves a mutation in the gene for the α1 subunit of the glycine receptor (21). However, the cost and inconsistent availability of these animals precludes their routine use for developing highly specific antimyoclonic drugs. Economic forces continue to drive the development of AEDs for partial seizures, with subsequent adoption of some AEDs to treat myoclonic seizures based on clinical experience.
Treatment Options for Specific Epilepsy Syndromes with Myoclonic Seizures in Early Infancy
Wheless and Sankar, recently reviewed this topic in detail (22). In this chapter, we limit our discussion to the treatment of those disorders that generally have their onset in the first 5 years or so of life. Detailed discussion of the pharmacokinetic and pharmacodynamic properties are not considered within the scope of this discussion. We have preferred to indicate the available choice of therapeutic agents and provide the literature citations to support the basis for that choice. Detailed and specific guidelines on the use of specific medications can be obtained elsewhere.
The information provided in this chapter on the treatment options for clinical syndromes involving myoclonus as a prominent feature (with the exception of the stiripentol trial) is not derived from controlled clinical trials. It remains difficult to quantify myoclonic seizures accurately to assess the effect of treatment precisely. Fortunately, myoclonic seizures are often associated with more easily quantified seizures such as astatic drop attacks, absence seizures, generalized clonic and generalized tonic–clonic seizures (GTCS). Further, many of these syndromes are relatively less common, compared to localization-related partial epilepsies with or without secondary generalization. In this chapter, we provide the guidelines in the management of the relatively more common epilepsies of infancy and early childhood, the Dravet syndrome of SMEI (Fig. 23-1) and genetic form of myoclonic-astatic epilepsy (MAE) (Fig. 23-2). Much of the information in these algorithms are obtained from what the American Academy of Neurology Quality Standards Subcommittee practice guidelines (23) would categorize as Class II (case-controlled and cohort studies) and Class III (case series, case reports, and expert opinions) evidence, rather than Class I (controlled clinical trials) evidence.
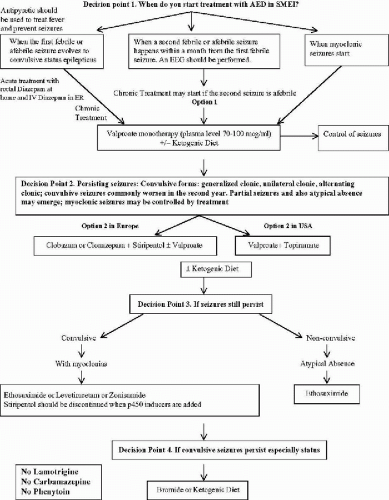 FIG. 23-1. Severe myoclonic epilepsy of infancy (Dravet Syndrome). |
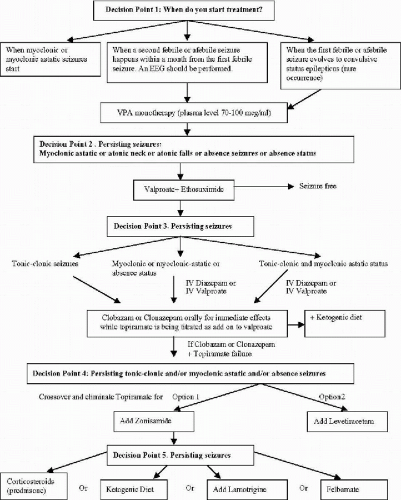 FIG. 23-2. Idiopathic or genetic myoclonic-astatic of early childhood. |
The algorithms represent practice habits of the experts who have the most extensive experience in these syndromes, these practice habits should be put to a test by controlled clinical trials.
Early Myoclonic Encephalopathy
Massive bilateral or axial myoclonias are the myoclonic features of early myoclonic encephalopathy. This rare syndrome has its onset during the first month of life and is often fatal by 1 year of age. The electroencephalogram (EEG) in patients with this condition reveals a burst-suppression pattern and may evolve toward typical hypsarrhythmia (24). No controlled clinical trials have investigated the treatment of early myoclonic encephalopathy. Treatment options are all based on anecdotal reports, and although drug therapy has demonstrated some efficacy, response to treatment is typically poor, and seizure freedom is rarely achieved. None of the conventional AEDs, adrenocorticotropic hormone (ACTH) gel, prednisone, or pyridoxine has been effective in treating this syndrome (25). The myoclonias of this syndrome gradually decrease with age. However, the overall prognosis is poor, and patients evolve towards West syndrome and eventually towards Lennox–Gastaut syndrome (24).
Infantile Spasms—West Syndrome
The treatment of IS is based on the underlying etiology of the spasms (25,26). With respect to both seizure control and developmental outcome, infants who have underlying focal cortical dysplasia, porencephalic cysts, or (rarely) a brain tumor may show the best response to epilepsy surgery. At UCLA such surgery is undertaken early in the course of the syndrome with the goal of maximizing the developmental outcome. Both anecdotal and controlled trial data indicate that vigabatrin is the treatment of choice for IS associated with tuberous sclerosis complex (28,29,30,31,32). Children with cryptogenic IS show the best response to ACTH in clinical trials (27,33). Use of VGB (27,28,29,30,34) and nitrazepam (35) to treat cryptogenic IS has also been evaluated in clinical trials. In Japan, pyridoxine is typically administered as initial therapy, but has a low response rate (36,37,38). Other treatment options that have been pursued in case series include VPA, prednisone, felbamate, LTG, TPM, and zonisamide (ZNS) (33,34,39,40). The ketogenic diet has recently shown success in children with refractory IS (cryptogenic and symptomatic etiologies) (41). Children with symptomatic etiologies are often treated with the same medications, but the response rate is much lower than in children with a cryptogenic etiology (27,33,34,39). As such, in patients with symptomatic etiologies, a risk/benefit assessment must be made for each therapeutic option and discussed with the child’s parents.
Related posts:
 Progressive Myoclonus Epilepsies: EPM1, EPM2A, EPM2B
Progressive Myoclonus Epilepsies: EPM1, EPM2A, EPM2B
 Myoclonic Status in Nonprogressive Encephalopathies
Myoclonic Status in Nonprogressive Encephalopathies
 Myoclonic–Astatic Epilepsy of Early Childhood – Definition, Course, Nosography, and Genetics
Genetics of Juvenile Myoclonic Epilepsy: Faulty Components and Faulty Wiring?
Ketogenic Diet in Patients with Dravet Syndrome and Myoclonic Epilepsies in Infancy and Early Childhood
Myoclonic–Astatic Epilepsy of Early Childhood – Definition, Course, Nosography, and Genetics
Genetics of Juvenile Myoclonic Epilepsy: Faulty Components and Faulty Wiring?
Ketogenic Diet in Patients with Dravet Syndrome and Myoclonic Epilepsies in Infancy and Early Childhood
 Treatment of Myoclonic Epilepsies of Childhood, Adolescence, and Adulthood
Treatment of Myoclonic Epilepsies of Childhood, Adolescence, and Adulthood
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


