Trimethadione, Paraldehyde, Phenacemide, Bromides, Sulthiame, Acetazolamide, and Methsuximide
Thomas R. Browne
Barbara W. LeDuc
Maria D. Kosta-Rokosz
Edward B. Bromfield
R. Eugene Ramsay
John De Toledo
Introduction
Trimethadione, paraldehyde, phenacemide, bromides, sulthiame, acetazolamide, and methsuximide are antiepileptic drugs (AEDs) that are rarely used today, because for most patients they are generally less effective or more toxic than alternative drugs. Each of these, however, may still occasionally be useful in carefully selected patients or special situations. Only paraldehyde and acetazolamide are available in the United States.
Trimethadione
Trimethadione is not marketed in the United States.
Structure and Chemistry
Trimethadione (3,5,5-trimethyl-2,4-oxazolidinedione; Tridione; TMO) is an oxazolidine-dione with a ring structure similar to that of other classes of antiepileptic drugs (Fig. 1). Several other oxazolidinediones also possess antiepileptic properties. Dimethadione (DMO) is of interest because it is the major, and perhaps the only, metabolite of TMO. Both TMO and DMO are white crystalline powders with molecular weights of 143.15 and 129.12, respectively. Trimethadione is readily soluble in aqueous and organic solvents.
Basic Mechanism of Action
A number of pharmacologic agents, including pentylenetetrazol, γ-hydroxybutyrate, and penicillin, induce seizures in rodents with clinical and EEG manifestations that are very similar to those of human absence seizures.203 Such models are reproducible and easy to standardize, and they are useful in predicting the antiabsence activity of a drug in humans. Ethosuximide and DMO block the seizure manifestations of this group of agents.
T-type calcium channels (TCCs) are present in high densities in thalamic neurons and contribute importantly to the regenerative bursts that drive normal and pathologic thalamocortical rhythms.45,46,47,48 Ethosuximide and DMO at therapeutic concentrations reduce TCCs when applied to thalamic neurons, whereas phenytoin and carbamazepine do not.45,48 Reduction of TCCs is probably the mechanism of antiabsence activity for DMO.15,45,175 Dimethadione may decrease the number of available TCCs or reduce individual TCC calcium conductance.45
Pharmacologic Fundamentals
Absorption and Routes of Administration
Trimethadione is rapidly absorbed by the oral route. Peak plasma TMO concentrations occur 30 to 60 minutes after oral administration.246
Distribution and Protein Binding
Neither TMO nor DMO binds to plasma proteins.24,246 Trimethadione is distributed to the total body water in humans24,246 and, in animals, has highest concentrations in brain, kidney, muscle, and liver.247 Animal work indicates that DMO is rapidly distributed to, but not bound by, the various body tissues and has a volume of distribution somewhere between extracellular water and total body water volume.247
Metabolism, Including Active Metabolites, and Excretion
Trimethadione is quantitatively demethylated to DMO by he-patic cytochrome P450 enzymes (probably isoforms CYP3A4, CYP2C9, and CYP2E1).34,76,166,216,249 Dimethadione is the only important metabolic product of TMO247 and does not undergo further metabolic change. Of an administered dose of TMO, 96% to 99% is excreted in the urine as DMO (trimethadione and DMO have approximately equal antiepileptic properties).24,75,245 Because TMO is rapidly metabolized to DMO, which is slowly excreted by the kidneys, DMO is the major antiepileptic drug present in the plasma of patients on chronic oral TMO therapy. The ratio of TMO to DMO in the plasma during chronic oral TMO therapy is approximately 1:20.24,25
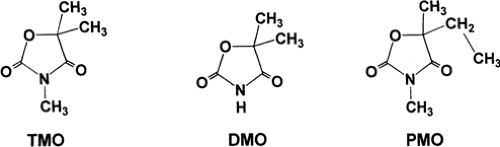 FIGURE 1. Structural formulas of trimethadione (TMO), dimethadione (DMO), and paramethadione (PMO). |
Clinical Pharmacokinetics
The elimination half-life of TMO is approximately 12 to 16 hours in the young and 24 hours in the elderly.24,25,76,215 The resulting DMO is excreted very slowly, with an elimination half-life of approximately 10 days or more.25,35,76,115 The very slow elimination of DMO has three clinical consequences. First, the time for buildup to a steady-state DMO level is 30 days or more, and therefore, it takes many days before the full therapeutic (and toxic) effects of a given dose of TMO are realized.38,89,246 Second, cessation of TMO therapy does not result in an immediate drop in DMO plasma concentration or in an immediate increase in seizure frequency.89 Third, DMO
plasma concentrations vary very little during the day, whereas plasma concentrations of TMO (which is more rapidly cleared) vary by 30% between peak and trough values on a three- or four-dose daily regimen.25
plasma concentrations vary very little during the day, whereas plasma concentrations of TMO (which is more rapidly cleared) vary by 30% between peak and trough values on a three- or four-dose daily regimen.25
Several studies have indicated that 700 μg/mL is the lower limit of the therapeutic range for plasma DMO level.23,25,38,115 The majority of patients with absence seizures who have DMO levels above 700 μg/mL have good control of seizures, whereas most patients with DMO levels below 700 μg/mL do not.25
Adverse Effects
Dose-related Effects
Hemeralopia (day blindness) is a phenomenon in which visual acuity is normal in low illumination but decreases when illumination is normal or brighter than normal. This is thought to be a retinal phenomenon in which the neuronal elements of cones (but not photochemical elements) are affected.202 There are no external or ophthalmoscopic changes.134 Detailed study reveals that TMO greatly prolongs the time of visual acuity adaptation when illumination changes from low to high or from high to low.202 In most patients, visual acuity becomes normal after adaptation is complete.202 Hemeralopia occurs in 30% of patients taking TMO133,236 and disappears 1 to 10 weeks after TMO is discontinued.78,133
Drowsiness occurs in 3% to 6% of patients on TMO133,236 but often subsides as tolerance to the drug develops.78 Behavioral disturbances occur in 4% to 8% of patients on TMO.133,236 Other central nervous system (CNS) side effects may include malaise, insomnia, vertigo, headache, paresthesias, and hiccups.25,78,133,236 Increased frequency of tonic–clonic seizures has been reported by some workers82 and denied by others.54,177 Nausea, vomiting, abdominal pain, or gastric distress occurs in 7% to 8% of patients on TMO.133,236
Idiosyncratic Effects
Hematologic side effects are the most frequent and troublesome serious side effects of TMO.78,236 Three classes of adverse hematologic responses to TMO have been identified: Modified normal response, controlled neutropenia, and pancytopenia.78 More than 3,000 neutrophils per cubic millimeter constitutes a modified normal response, whereas less than this number is called a controlled neutropenia. The incidence of controlled neutropenia in association with TMO therapy is about 20%.51,236 Unfortunately, the peripheral blood count is not an accurate reflection of changes in the bone marrow, and agranulocytosis may be established in the bone marrow well before the peripheral count is altered.57 There have been at least 19 reported cases of pancytopenia in association with TMO therapy, and 13 ended fatally.236 The earliest change in the peripheral blood is a decrease in megakaryocytes, followed by a reduction in the number of platelets; this in turn is followed by a prolonged clot retraction time.57 Frequent examination of peripheral blood, including megakaryocytes and platelets, and measurements of clot retraction time are essential in patients taking TMO.
A possible relationship between TMO and lymphadenopathy or tumors of blood-forming organs has been discussed by Gallagher.78 Firm proof of such a relationship has not been established.78
Dermatologic side effects include rash, erythema multiforme, and exfoliative dermatitis (including one fatal case)79,236 and occur in 9% to 14% of patients taking TMO.133,236 These disorders usually occur early in the course of TMO therapy and may be more frequent in children under the age of 10 years.133 The more severe dermatologic reactions are rare and are generally nonfatal.78
At least nine cases of nephrotic syndrome (albuminuria, decreased plasma albumin, hypercholesterolemia) in association with TMO therapy have been reported.78,236 Two of these cases ended fatally.236
Teratogenicity
There are numerous reports of fetal malformations in association with TMO therapy.26,86,164,216 A large collaborative study reported malformations in 18 of 61 children born to mothers who had taken TMO during the first trimester.165 In some children, a characteristic set of findings has been termed the “fetal trimethadione syndrome.”26,86 Malformed or low-set ears, cleft lip and palate, delayed mental development, speech impairment, urogenital malformations, V-shaped eyebrows, irregular teeth, skeletal malformations, and cardiac defects are the common features.26,86 Less common features of the fetal trimethadione syndrome include intrauterine growth retardation, short stature, microcephaly, ocular anomalies, and simian creases.86 In addition, the spontaneous abortion rate is high.26 The mechanism of TMO teratogenicity may be oxidative macromolecular damage.237 Although the mother was taking other drugs in most of these cases, the evidence suggesting TMO teratogenesis is so strong that TMO should be given during pregnancy only if the potential benefits are great enough to outweigh the considerable potential risks.
Drug Interactions
Although TMO and DMO have been in clinical use for many years, reports of interactions with other drugs are scarce.248 The following animal data may have some clinical relevance: (a) TMO and methobarbital interfere with demethylation of each other by liver microsomes; (b) DMO inhibits demethylation of TMO and methobarbital; (c) drugs causing a distortion of acid–base balance (e.g., NH4Cl, acetazolamide, and NaHCO3) can affect the distribution and excretion of DMO (a weak acid); and (d) TMO does not induce liver enzymes.
The Role of the Drug
Trimethadione is no longer manufactured in the United States. Chewable tablets (150 mg) are manufactured in Europe (Slovakofarma AS).
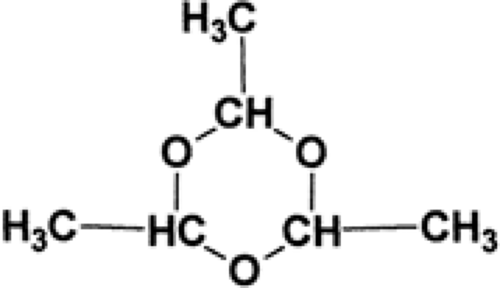 FIGURE 2. Structural formula of paraldhyde. |
Trimethadione has been used to treat absence seizures that are refractory to less toxic drugs (i.e., ethosuximide, valproic acid, clonazepam, acetazolamide, and lamotrigine). Millichap and Aymat159 reviewed 431 reported cases of absence seizures treated with TMO. Absence seizures were reduced in frequency by 75% or more in 56% of cases and were completely controlled in 18% of cases. Coatsworth’s review44 showed similar results. The drug thus possesses considerable antiabsence activity. A trial of TMO may be indicated when absence seizures are not controlled with less toxic drugs and when the potential benefits of TMO outweigh the potential risks (see above). Trimethadione is also sometimes effective for treating the refractory atypical absence, myoclonic, and atonic seizures of the Lennox-Gastaut syndrome.52,56,134 TMO is also used as a cytochrome P450 2E1 probe for hepatic disease.166,217
How to Use the Drug
The usual starting daily dose is 0.9 g in adults and 0.3 g in children in three or four divided doses. Because of the long elimination half-life of DMO, adequate plasma concentrations should be maintained with doses given once daily, although such a regimen has not been systematically studied to determine the plasma concentrations and side effects of once-daily administration. The dosage of TMO may be increased at weekly intervals by 150 mg/day in children and 300 mg/day in adults. Maintenance dosage should be the smallest amount of drug required to maintain adequate seizure control. An oral solution form is available for children.
Paraldehyde
Structure and Chemistry
Paraldehyde (2,4,5-trimethyl-1,3,5-trioxane; PARALDEHYDE) is a cyclic polymer of acetaldehyde (Fig. 2). It is a colorless liquid with a strong aromatic odor and a burning, disagreeable taste. It is miscible with oils and has a molecular weight of 132.16.
Basic Mechanism of Action
The mechanism of action of paraldehyde is unknown.
Pharmacologic Fundamentals
Absorption and Routes of Administration
The times to peak plasma concentration of paraldehyde by various routes are as follows: IV route, immediately after infusion79; IM route, 20 to 60 minutes1; oral route (in water), 30 minutes3; oral route (in oil), 2 to 4 hours79; rectal route (in oil), 2 to 4 hours.8,79 Clinical effects are usually evident long before maximum plasma concentration.8,79,193,224
Distribution
Paraldehyde is rapidly distributed to the brain. Following an IV injection, drowsiness ensues within 2 to 5 seconds, and anesthesia occurs within <2 minutes.8,79,193 The steady-state volume of distribution of paraldehyde is 890 mL/kg.8 Paraldehyde readily crosses the placental barrier and may cause delayed respirations in neonates.79
Metabolism and Excretion
Seventy to eighty percent of paraldehyde is metabolized by the liver, 20% to 30% is exhaled by the lungs, and a very small amount is excreted unchanged by the kidney.14 In patients with liver disease, the rate of elimination of paraldehyde and the percentage of paraldehyde eliminated by the liver decrease, and the percentage of paraldehyde eliminated by the lungs increases.
Elimination Half-Life
The elimination half-life of paraldehyde in adults and children is 3.4 to 9.8 (mean 6.1 to 7.4) hours.6,224 In two studies performed in neonates, the elimination half-life of paraldehyde was 10.2 ± 1.0 and 18.1 ± 5.5 hours.79,83,90 The plasma disposition kinetics of paraldehyde fit an open two-compartment model after intravenous injection.8 The elimination half-life of paraldehyde is prolonged in patients who have received phenobarbital before paraldehyde.79
Adverse Effects
Since paraldehyde was introduced in 1882, there have been at least 95 reports of death associated with its use.14,103 These reports have led to widespread disrepute and infrequent use of paraldehyde. However, many (probably most) of these deaths were from one of the following factors: (a) suicidal overdosage, (b) use of decomposed drug, (c) use of doses larger than those currently recommended, and (d) use of improperly diluted paraldehyde. The incidence of death and serious side effects with U.S. Pharmacopeia (USP)-quality paraldehyde at recommended doses and following recommended administration procedures is unknown but probably is less than is generally supposed.
Overdosage
Side Effects
By Any Route
Paraldehyde administered by any route may cause disagreeable breath odor (from exhaled paraldehyde), right heart failure, pulmonary edema and hemorrhage (especially if the drug is used in excessive dosage), rash, irritability, toxic hepatitis, or toxic nephritis.14,34,201 Chronic use of paraldehyde may produce a metabolic acidosis.103 Chronic paraldehyde use may
produce tolerance or physical dependence with a withdrawal syndrome similar to that seen with alcohol withdrawal.107
produce tolerance or physical dependence with a withdrawal syndrome similar to that seen with alcohol withdrawal.107
Intravenous Route
Three series18,79,193 (totaling 181 patients) in which IV paraldehyde was used as an anesthetic agent report no serious complications, although many patients experienced coughing, choking, pharyngeal irritation, tachycardia, or pain at the site of infusion. On the other hand, there are case reports of IV paraldehyde suddenly producing apnea, coughing, cyanosis, hypotension, and clinical and radiographic signs of pulmonary edema.201 All the above studies involved injection of undiluted or 10% solution of paraldehyde and thus failed to properly dilute paraldehyde so that it would remain soluble after IV injection (see below). Gessner82 postulates that the above-cited pulmonary–cardiac complications were the result of pulmonary embolism of precipitated droplets of pure paraldehyde in the bloodstream as a result of improper dilution and cites two studies8 in which properly diluted paraldehyde (4% to 8% solution) was administered IV without complications. Note that intra-arterial injection of paraldehyde may cause vascular injury96,234 and that direct injection of undiluted paraldehyde into an umbilical artery catheter may cause vascular injury and injury to tissues by microembolization.90,97
Oral and Rectal Routes
Paraldehyde can cause irritation and corrosion of the mouth and stomach when given orally and of the rectum and large intestine when given rectally.1 Rectal administration of decomposed paraldehyde has resulted in perforation of the large intestine.1 The drug should be diluted properly before administration by these routes (see below).
Intramuscular Route
Role of the Drug
Status Epilepticus
Evidence for the efficacy of paraldehyde in status epilepticus consists of uncontrolled trials2,29,98,154,241,244 and testimo-nials.29,55,238 One open-label study found that intramuscular paraldehyde controlled seizures lasting more than 5 minutes in children within 10 minutes of administration in 49% of patients.2 These reports contain few details but suggest that paraldehyde can probably control status epilepticus in most adults and children; may control status epilepticus when other agents fail; and may be more effective for tonic–clonic status epilepticus than for partial status epilepticus. On the whole, paraldehyde is disappearing from hospital pharmacies as better and easier-to-store antiepileptic drugs have become widely available in the United States. However, paraldehyde is still used to treat status epilepticus in developing countries.2 Comparison of the reported evidence leads to the conclusion that for almost all patients, IV lorazepam or diazepam is preferable to IV paraldehyde when immediate control of seizures is necessary, and that for most patients a loading dose of phenytoin or phenobarbital followed by maintenance doses is preferable to continued paraldehyde.32,113 In patients with tonic–clonic status epilepticus refractory to first-line drugs (e.g., lorazepam, diazepam, phenytoin, and phenobarbital), anesthetic coma (if available) is probably more effective and safer than paraldehyde. There are three special situations in which paraldehyde may still find limited used for status epilepticus: (a) when the initial therapy must be given IM; (b) when the patient is allergic to safer agents; and (c) when phenytoin, phenobarbital, lorazepam, and diazepam fail, and anesthetic coma cannot be practically managed (no intensive care unit [ICU], no ventilator).
Paraldehyde may be the drug of choice for status epilepticus when drugs can be administered only by the IM route (e.g., no physician immediately available, suitable vein cannot be found, resuscitation equipment not available). Paraldehyde absorption by the IM route produces near-peak plasma concentrations in 15 to 20 minutes. Lorazepam, diazepam, phe-nytoin, and phenobarbital require significantly longer times for absorption by the IM route and do not produce therapeutic plasma concentrations within 15 to 20 minutes when given in the usual doses IM.105,110,127,210,232,240
Patients with no history of seizures except during alcohol withdrawal and patients with true seizure disorders and therapeutic plasma concentrations of antiepileptic drugs who have seizures only when withdrawing from alcohol are most often treated with benzodiazepines. Paraldehyde is an effective alternative to benzodiazepines, but paraldehyde probably has more side effects.29,203 The preponderance of experimental and clinical evidence suggests that phenytoin is not effective for alcohol withdrawal seizures (see Chapter 268), and barbiturates are not generally used for treating alcohol withdrawal symptoms in the United States because of concern about habituation.
When status epilepticus continues after full loading doses of phenytoin and phenobarbital have been administered, paraldehyde is sometimes effective in stopping seizures.29,238 However, the greater certainty of seizure control and greater safety of anesthetic coma makes it the treatment of choice, if available, for refractory status epilepticus.
Alcohol Withdrawal Syndrome and Delirium Tremens
Other Indications
How to Use the Drug
Formulation
The water solubility of paraldehyde is greatest (12.8%) at 12°C and decreases as the temperature rises above, or falls below, this point. At 37°C, the water solubility of paraldehyde is 7.8%. Lack of awareness of these critical facts has led to the practice of injecting paraldehyde intravenously in its pure form or as a 10% solution. In either case, the solubility of paraldehyde at 37°C would be exceeded, and droplets of pure paraldehyde may form in the bloodstream, which can result in pulmonary emboli (see above).
Paraldehyde has a slight tendency to depolymerize back to acetaldehyde. In the presence of air, acetaldehyde oxidizes to acetic acid, which then acts as a catalyst for further depolymerization of paraldehyde to acetaldehyde. Improper storage of paraldehyde has resulted in some samples containing 40% to 98% acetic acid,103 and as little as 7 mL of such decomposed paraldehyde has proved fatal.6 A survey of 42 paraldehyde samples collected from hospital wards in 1957 revealed that only 11% met USP standards.103
In 1965, USP specifications were altered to state that paraldehyde must be preserved “in well-fitted, tight, light-resistant containers not exceeding 30 ml and that the unused contents of any container that has been opened more than 24 hr be discarded.” Such procedures should reduce (but may not completely eliminate) the hazards of decomposed paraldehyde.
Treatment of Status Epilepticus
The dosage of paraldehyde for treatment of status epilepticus is 0.1 to 0.15 mL/kg. This dose may be repeated every 2 to 4 hours if necessary. Before any paraldehyde is administered, it should be checked for purity and conformity with USP standards for storage.29 The minimum therapeutic plasma concentration of paraldehyde for control of status epilepticus is approximately 300 ng/mL.98
The safety of paraldehyde by the IV route is controversial. When immediate control of seizures with an IV medication is indicated, one should probably use another drug (e.g., lorazepam or diazepam) whose safety by the IV route is much greater. If IV paraldehyde must be given, it needs to be diluted to a 4% solution and infused slowly.
Paraldehyde decomposes many plastics, including poly-styrene and styrene–acrylonitrile copolymer, and also rubber.29,69,81,116,194,199,239 Glass syringes usually are recomm-ended.118
Absorption by way of the oral route is slower than by the IM route.29 There is also a risk of aspiration of paraldehyde, which is highly noxious to the lungs. The oral route is best avoided in patients with status epilepticus.
The ease of administration of paraldehyde by way of the rectal route probably accounts for its frequent administration this way. However, rectal absorption is considerably slower than that by the IM or oral route, making the rectal route particularly undesirable for treating status epilepticus.75 Furthermore, the slow rectal absorption of paraldehyde can result in administration of large doses to obtain a plasma concentration high enough to control seizures. When the large rectal reservoir of paraldehyde is eventually absorbed, toxic plasma concentrations of paraldehyde may result.31 If rectal paraldehyde is administered, it should be diluted 2:1 in oil (olive or cottonseed) or diluted in 200 mL of 0.9% sodium chloride.
Phenacemide
Phenacemide is not marketed in the United States.
Structure and Chemistry
Phenacemide (phenylacetylurea, phenacetylcarbamide, Phe-nurone, PAC) is a straight-chain analog of 5-phenylhydantoin (Fig. 3).
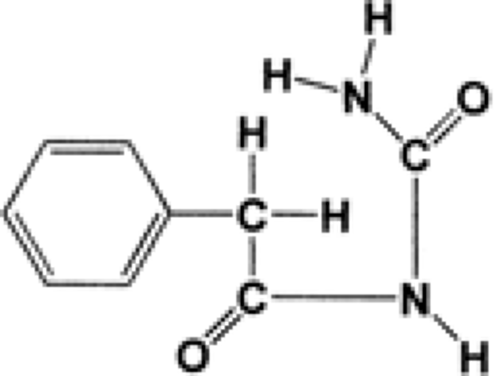 FIGURE 3. Structural formula of phenacemide. |
Table 1 Toxicity of Phenacemide* | ||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||
Basic Mechanism of Action
In experimental models of epilepsy, phenacemide elevates the seizure threshold for minimal electroshock, maximal electroshock, and pentylenetetrazol seizures.158,212 In fact, phenacemide has higher protective indexes against minimal electroshock seizures (a model of complex partial seizures) and maximal electroshock seizures (a model of tonic–clonic seizures) than either phenytoin or phenobarbital.212 The mechanism of action of phenacemide is probably similar to that of phenytoin because of the similar structural and three-dimensional conformation of phenacemide and phenytoin.36,249
Pharmacologic Fundamentals
Phenacemide is well absorbed from the intestine, with peak plasma concentrations occurring 3 to 5 hours after a single oral dose.229 The extent of protein binding is unknown. Phenacemide is metabolized by means of hepatic microsomal enzymes by para-hydroxylation of the phenyl group followed by conjugation to a glucuronic acid.193,229 Ring closure to form hydantoin does not occur.31 The metabolite and its glucuronide are excreted through the kidneys, with little or no unchanged phenacemide found in the urine.31 The elimination half-life of phenacemide is 5 to 12 hours.31,192 The effective plasma concentration ranges from 16 to 75 μg/mL.229
Adverse Effects
The frequencies of the most common side effects of phenacemide are summarized in Table 1.
Dose-related Effects
Anorexia, nausea, vomiting, weight loss, and abdominal pain occur in 8% to 13% of patients on phenacemide.89,140,227 In about 3% of such cases, the drug must be discontinued.142 Headache or a feeling of “fullness in the head” occurs in 2% to 6% of patients on phenacemide.49,89,140,227 The headache appears during the first few days of phenacemide therapy and is often severe. Drowsiness occurs in up to 5% of patients.227
Idiosyncratic Effects
Destructiveness, belligerence, marked irritability, restlessness, and depression are the most commonly observed behavioral changes.89,140,227 Suicidal tendencies, paranoia, and acute psychotic states have also been reported.227 Behavioral symptoms occur most commonly during the first 4 to 6 weeks of phenacemide treatment and disappear within a few weeks of stopping the drug.134,227 Behavioral or psychiatric changes necessitate discontinuing phenacemide in about 10% of patients.227 On the other hand, in some patients with pre-existing personality disorders, improvement has been noted after starting treatment with phenacemide.49,140 In patients who have a history of psychiatric disorders, hospitalization during the first week of phenacemide therapy may be advisable. Patients and family should be alerted to report changes in behavior such as decreased interest in surroundings, depression, or aggressiveness.
Rashes of the maculopapular, scarlatiniform, or acneiform type may occur.40,134,140 Rashes are most common during the first 3 weeks of phenacemide therapy and may be accompanied by fever.49,134 The rashes are not dose related and disappear within a week of stopping the drug.49
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


