Chapter 51 Neurophysiology of Seizures and Epilepsy
A seizure is defined as abnormal neuronal firing leading to a clinical alteration of neurologic function (motor, sensory, autonomic, or psychological). Electrical activity underlying a seizure is the net product of biochemical processes at the cellular level occurring in the context of large neuronal networks and likely involves several key cortical and subcortical structures. The output of this activity is reflected on the surface electroencephalogram (EEG), which is the primary clinical tool for measuring normal and abnormal brain electrical activity. Epilepsy is the condition of recurrent spontaneous seizures arising from aberrant electrical activity within the brain. Epilepsy is not a singular disease, but rather is heterogeneous in terms of clinical expression, underlying causes, and pathophysiology (Table 51-1). An epilepsy syndrome refers to a group of signs and symptoms that usually occur together, such as seizure type, age of seizure onset, responsiveness to a particular AED, and characteristic EEG findings, genetics, and natural history. Epileptogenesis is the process by which neural circuits undergo structural or physiological changes, resulting in an enduring epileptic state.
Table 51-1 Examples of Pathophysiologic Defects Leading to Epilepsy
| Level of Brain Function | Condition | Pathophysiologic Mechanism |
|---|---|---|
| Neuronal network | Cerebral dysgenesis, post-traumatic scar, mesial temporal sclerosis (in TLE) | Altered neuronal circuits; formation of aberrant excitatory connections (i.e., sprouting) |
| Neuron structure | Down syndrome and other syndromes with mental retardation and seizures | Abnormal structure of dendrites and dendritic spines; altered current flow in neuron |
| Neurotransmitter synthesis | Pyridoxine (vitamin B6) dependency | Decreased GABA synthesis; B6, a co-factor for GAD |
| Neurotransmitter receptors, inhibitory | Angelman syndrome, juvenile myoclonic epilepsy | Abnormal GABA receptor subunits |
| Neurotransmitter receptors, excitatory | Nonketotic hyperglycinemia | Excess glycine leads to activation of NMDA receptors |
| Synapse development | Neonatal seizures | Many possible mechanisms, including the depolarizing action of GABA early in development |
| Ion channels (channelopathies) | Benign familial neonatal convulsions Dravet syndrome | Potassium channel mutations Sodium channel mutations |
GABA, gamma-aminobutyric acid; GAD, glutamic acid decarboxylase; NMDA, N-methyl-d-aspartate; TLE, temporal lobe epilepsy.
Classification of Seizures
Seizures are broadly divided into two groups, depending on their site of origin and pattern of spread (Figure 51-1). Partial seizures arise from a localized region of the brain, and the associated clinical manifestations are related to the function ordinarily subserved by that area. Focal discharges can spread locally through synaptic and nonsynaptic mechanisms, distally to subcortical structures, and through commissural pathways that may eventually involve the entire cortex; this evolution is believed to occur when focal seizures secondarily generalize. For example, a seizure arising from the left motor cortex may cause jerking movements of the right upper extremity. If epileptiform discharges subsequently spread to adjacent areas and eventually encompass the entire brain, it is described as a secondarily generalized tonic-clonic seizure.
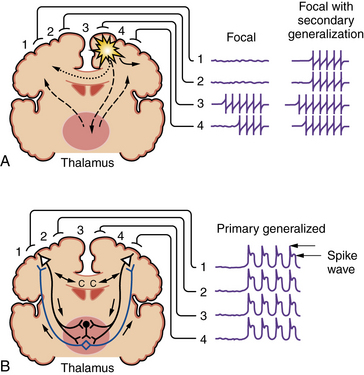
Fig. 51-1 Coronal brain sections depicting seizure types and potential routes of seizure spread.
(From Stafstrom CE. An introduction to seizures and epilepsy: Cellular mechanisms underlying classification and treatment. In: Stafstrom CE, Rho JM, eds. Epilepsy and the ketogenic diet. Totowa, NJ: Humana Press, 2004;3–29. Reprinted with kind permission of Springer Science+Business Media.)
Primary generalized seizures begin with abnormal electrical discharges in both hemispheres simultaneously and involve reciprocal thalamocortical connections (see Figure 51-1). The EEG signature of a primary generalized seizure is bilateral synchronous spike-wave discharges seen across all scalp electrodes. The manifestations of such widespread epileptiform activity can range from brief impairment of consciousness (e.g., an absence seizure) to rhythmic jerking movements of all extremities accompanied by loss of posture and consciousness.
Epilepsies or epilepsy syndromes have likewise been categorized into those in which seizures begin focally (partially) or throughout the brain (generalized), with further division into those that have a known etiology (symptomatic) and those that do not (idiopathic; many of these have a genetic basis) [Commission, 1989]. The classification of epilepsy syndromes is undergoing revision as new knowledge of epilepsy genetics and pathophysiology emerges [Engel, 2006].
Cellular Electrophysiology
Excitation/Inhibition Balance
Although there are differences in the mechanisms that underlie partial and generalized seizures, it is useful to view any seizure activity as a perturbation in the normal balance between inhibition and excitation. This imbalance can involve a localized region, multiple brain areas, or the entire brain, and often reflects a combination of increased excitation and decreased inhibition. However, the traditional concept of a seizure arising when the excitation/inhibition balance is altered is useful but oversimplified. It is now appreciated that factors once thought to be solely inhibitory (e.g., gamma-aminobutyric acid (GABA) synaptic transmission) can actually be excitatory in some circumstances (see Increased Seizure Susceptibility of the Immature Brain, below). Furthermore, synaptic inhibition is critical for certain normal brain rhythms and probably also plays a role in abnormal rhythmic activity, such as spike-wave discharges [Fritschy, 2008].
Structural Correlates of Epilepsy: Hippocampus and Neocortex
Brain regions differ in their propensity to generate seizures, based on intrinsic membrane properties, synaptic organization, cell density, and pattern of cellular inteconnectivity. Even within the same brain region, cell types differ with regard to their excitability [Steriade, 2004]. The hippocampus and neocortex are particularly prone to seizure generation. The hippocampus, with its orderly laminar organization and trisynaptic excitatory circuitry, has been used extensively in electrophysiologic studies of seizure mechanisms [Schwartzkroin and Mueller, 1987].
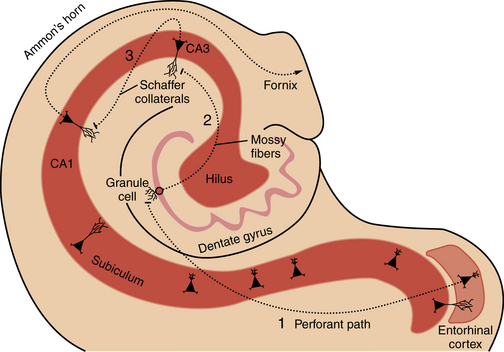
Familiarity with hippocampal anatomy will facilitate understanding of the physiological concepts discussed below (Figure 51-2). The hippocampal formation consists of the dentate gyrus, hippocampus proper (i.e., Ammon’s horn, with subregions CA1, CA2, and CA3), subiculum, and entorhinal cortex. These four regions are linked by prominent excitatory, largely unidirectional, feed-forward connections. The forward-projecting trisynaptic circuit begins with neurons in layer II of the entorhinal cortex that project axons to the dentate gyrus along the perforant pathway, where they synapse on granule cell and interneuron dendrites. Granule cells, the principal cell type of the dentate gyrus, send their axons, called mossy fibers, to synapse on cells in the hilus and in the CA3 field of Ammon’s horn. Several classes of inhibitory interneurons within the dentate hilus modulate on-going excitatory neuronal activity [Lawrence and McBain, 2003]. CA3 pyramidal cells project to other CA3 pyramidal cells through local collaterals, to the CA1 field of Ammon’s horn through Schaffer collaterals, and to the contralateral hippocampus. CA1 pyramidal cells send their axons into the subicular complex. Neurons of the subicular complex project to the entorhinal cortex and other cortical and subcortical targets.
Overview of Ion Channels
The key channels and receptors involved in normal and epileptic firing are summarized in Figure 51-3 and Table 51-2. Two major types of ion channels – voltage-gated and ligand-gated – are responsible for inhibitory and excitatory activity. Voltage-gated channels include sodium and calcium channels that function to depolarize the cell membrane toward the action potential threshold, while voltage-gated potassium channels largely dampen neural excitation. Voltage-gated channels are activated by membrane potential changes that alter the conformational state of the channel and allow selective passage of charged ions through a pore. Ligand-gated channels constitute the second type. In ligand-gated channels, a neurotransmitter (e.g., glutamate, GABA) is released from a presynaptic terminal (after presynaptic calcium influx) into the synaptic cleft and then binds with selective affinity to a membrane-bound receptor on the postsynaptic membrane. This binding activates a cascade of events, including a conformational shift to reveal an ion-permeant pore. Passage of ions across these channels results in depolarization (i.e., inward flux of cations) or hyperpolarization (i.e., inward flux of anions or outward flux of cations). Numerous ion channel mutations underlie epilepsy syndromes, giving rise to the concept of “epilepsy channelopathies” [Reid et al., 2009].
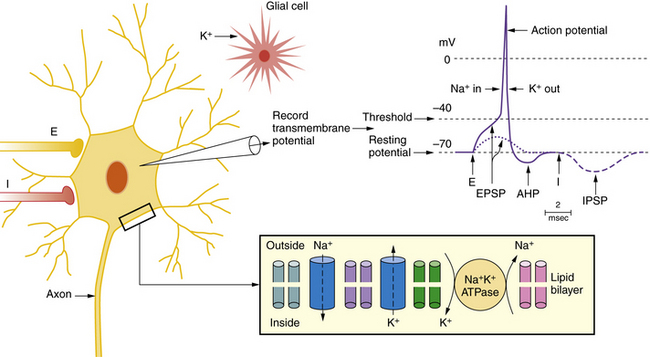
Table 51-2 Roles of Channels and Receptors in Normal and Epileptic Firing
| Channel or Receptor | Role in Normal Neuronal Function | Possible Role in Epilepsy |
|---|---|---|
| Voltage-gated Na+ channel | Subthreshold EPSP; action potential upstroke | Repetitive action potential firing |
| Voltage-gated K+ channel | Action potential downstroke | Abnormal action potential repolarization |
| Ca2+-dependent K+ channel | AHP after action potential; sets refractory period | Limits repetitive firing |
| Voltage-gated Ca2+ channel | Transmitter release; carries depolarizing charge from dendrites to soma | Excess transmitter release; activates pathophysiologic intracellular processes |
| Non-NMDA receptor (i.e., AMPA) | Fast EPSP | Initiates PDS |
| NMDA receptor | Prolonged, slow EPSP | Maintains PDS; Ca2+ activates pathophysiologic intracellular processes |
| GABAA receptor | IPSP | Limits excitation |
| GABAB receptor | Prolonged IPSP | Limits excitation |
| Electrical synapses | Ultrafast excitatory transmission | Synchronization of neuronal firing |
| Na+-K+ pump | Restores ionic balance | Prevents K+-induced depolarization |
AHP, after-hyperpolarization; AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; EPSP, excitatory postsynaptic potential; GABA, γ-aminobutyric acid; IPSP, inhibitory postsynaptic potential; NMDA, N-methyl-d-aspartate; PDS, paroxysmal depolarization shift.
Voltage-Dependent Membrane Conductances
Depolarizing Conductances
A rapidly inactivating inward sodium conductance underlies the depolarizing (excitatory) phase of the action potential, and a non-inactivating, persistent sodium current can augment cell depolarization (e.g., produced by excitatory synaptic input) in the subthreshold voltage range; both are critical for regulation of neuronal firing and play a role in epilepsy [Stafstrom, 2007b; Ragsdale, 2008]. Many AEDs act in part through interactions with voltage-dependent sodium channels [Rogawski and Loscher, 2004]. Each sodium channel is composed of a complex of three polypeptide subunits: a major α subunit and two smaller β subunits that influence the kinetic properties of the α subunit. The shape of action potentials is determined by the types of α and β subunits present in an individual neuron [Catterall et al., 2005].
Neurons also display voltage-gated inward calcium conductances. Calcium currents underlie burst discharges in hippocampal CA3 neurons. Activation of voltage-dependent calcium channels contributes to the depolarizing phase of the action potential, and can affect neurotransmitter release, gene expression, and neuronal firing patterns. Several distinct subtypes of calcium channels are distinguished on the basis of electrophysiologic properties, pharmacologic profile, molecular structure, and cellular localization [Catterall et al., 2003]. The molecular structure of voltage-gated calcium channels is similar to that of sodium channels. Voltage-dependent calcium channels are hetero-oligomeric complexes containing a principal pore-forming α-1 subunit and one or more smaller subunits (α2, β, γ, δ) that are not obligatory for normal activity but modulate the kinetic properties of the channel.
Hyperpolarizing Conductances
Depolarizing sodium and calcium currents are counterbalanced by an array of voltage-dependent hyperpolarizing (inhibitory) currents, primarily mediated by potassium channels. Potassium channels represent the largest and most diverse family of voltage-gated ion channels, and they function to decrease neuronal excitation [Gutman et al., 2005]. The prototypic voltage-gated potassium channel is composed of four membrane-spanning α subunits and four regulatory β subunits that are assembled in an octameric complex to form an ion-selective pore. Potassium conductances include a leak conductance, which is a major determinant of the resting membrane potential; an inward rectifier (involving the flux of other ions), which is activated by hyperpolarization; a large set of delayed rectifiers, which are involved in the termination of action potentials and repolarization of the neuron’s membrane potential; an A-current, which helps to determine interspike interval and affects the rate of cell firing; an M-current, which is activated by cholinergic muscarinic agonists and affects resting membrane potential and cell firing rate; and a set of calcium-activated potassium conductances, which are sensitive to intracellular calcium concentration and affect cell firing rate and interburst interval. (Rectification refers to differences in conductance depending on the direction of ion flow through the channel; rectification can also result from blocking of the pore by other ions.)
Modulation or facilitation of hyperpolarizing conductances can be viewed as potentially antiepileptic, and some newer AEDs act directly on voltage-gated potassium channels [Wickenden, 2002]. For example, topiramate induces a steady membrane hyperpolarization mediated by a potassium conductance, and levetiracetam blocks sustained repetitive firing by paradoxically decreasing voltage-gated potassium currents. Retigabine, an opener of Kv7 subtype potassium channels, has broad efficacy in animal seizure models and enhances activation of KCNQ2 and KCNQ3 potassium channels [Miceli et al., 2008]. This finding is particularly intriguing, given that mutations in genes encoding these proteins have been linked to a rare form of inherited epilepsy, benign familial neonatal seizures [Singh et al., 2003].
Synaptic Physiology
Inhibitory Synaptic Transmission
GABA, the main inhibitory neurotransmitter in the mature mammalian central nervous system, is a neutral amino acid synthesized from glutamic acid by the rate-limiting enzyme glutamic acid decarboxylase. GABA released from axon terminals binds to at least two classes of receptors, GABAA and GABAB, which are found on almost all cortical neurons [Sieghart, 2006].
The GABAA receptor is a macromolecular receptor complex consisting of an ion pore and binding sites for agonists and a variety of allosteric modulators, such as benzodiazepines and barbiturates, each differentially affecting the kinetic properties of the receptor. The GABAA receptor is a heteropentameric complex composed of combinations of several polypeptide subunits arranged in topographic fashion to form an ion channel. This channel is selectively permeable to chloride and bicarbonate ions. Seven types of subunits (α, β, γ, δ, ε, π, ρ) have been described, each with one or more subtypes [Wafford et al., 2004]. Although several thousand receptor isoforms are possible from differential expression and assembly of various subunits, only a limited number of functional combinations is likely to exist in the brain. Most functional GABAA receptors follow the general motif of containing either α and β or α, β, and γ subunits with variable stoichiometry. Because individual subunits may be differentially sensitive to pharmacologic agents, GABAA receptor subunits represent potentially useful molecular targets for new AEDs [Macdonald and Kang, 2009].
Activation of GABAA receptors on the somata of mature neurons generally results in the influx of chloride ions and consequent membrane hyperpolarization, inhibiting cell firing. In neurons of the immature brain, however, GABAA receptor activation causes depolarization of the postsynaptic membrane [Staley, 2006a]. This reversal of the conventional GABAA effect reflects a reversed chloride electrochemical gradient, a consequence of the evolving expression of cation chloride co-transporters during development (see Development of Neurotransmitters, Receptors, and Transporters, below).
In addition to GABAA receptors, metabotropic GABAB receptors are located on postsynaptic membrane and presynaptic terminals [Bettler and Tiao, 2006]. GABAB receptors act through guanosine triphosphate (GTP)-binding proteins to control calcium or potassium conductances. Whereas GABAA receptors generate fast, high-conductance, inhibitory postsynaptic potentials close to the cell body, GABAB receptors on the postsynaptic membrane mediate slow, long-lasting, low-conductance inhibitory postsynaptic potentials, primarily in dendrites. Perhaps of greater functional significance, activation of GABAB receptors on axon terminals blocks neurotransmitter release. It is thought that GABAB receptors are associated with terminals that release GABA on to postsynaptic GABAA receptors. In such cases, activation of GABAB receptors reduces the amount of GABA released, resulting in disinhibition [Simeone et al., 2003]. Abnormalities of GABAergic function, including synthesis, synaptic release, receptor composition, trafficking or binding, and metabolism, can each lead to a hyperexcitable, epileptic state [Cossart et al., 2005; Macdonald and Kang, 2009].
Excitatory Synaptic Transmission
Glutamate, an excitatory amino acid, is the principal excitatory neurotransmitter of the mammalian central nervous system. Glutamatergic pathways are widespread throughout the brain, and excitatory amino acid activity is critical to normal brain development and activity-dependent synaptic plasticity [Simeone et al., 2004]. There are two broad classes of glutamate receptors – ionotropic and metabotropic. Ionotropic glutamate receptors are divided into N-methyl-d-aspartate (NMDA) and non-NMDA receptors, based on biophysical properties and pharmacologic profiles. Each subtype of glutamate receptor consists of a multimeric assembly of subunits that determine its distinct functional properties. An NMDA receptor consists of an NR1 subunit plus NR2A, NR2B, NR2C, NR2D, and/or NR3A.
The NMDA receptor contains a binding site for glutamate (or NMDA) and a recognition site for a variety of modulators (e.g., glycine, polyamines, MK-801, zinc). NMDA receptors also demonstrate voltage-dependent block by magnesium ions. When the membrane is depolarized and the magnesium block of the NMDA receptor is alleviated, activation of the NMDA receptor results in an influx of calcium and sodium ions. Calcium entry is central to the initiation of a number of second messenger pathways, such as stimulation of a variety of kinases that subsequently activate signal transduction cascades, leading to changes in transcriptional regulation. Activation of the NMDA receptor leads to generation of relatively slow and long-lasting excitatory postsynaptic potentials. These synaptic events contribute to epileptiform burst discharges, and NMDA receptor blockade results in the attenuation of bursting activity in many models of epileptiform activity [Kalia et al., 2008].
Non-NMDA ionotropic receptors are divided into α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and kainate receptors [Vincent and Mulle, 2009]. AMPA receptors are composed of combinations of GluR1, GluR2, GluR3, and/or GluR4 subunits, while kainate receptors are composed of combinations of GluR5, GluR6, GluR7, KA1, and/or KA2 subunits. AMPA receptors are responsible for the major part of the excitatory postsynaptic potential – fast-rising and brief in duration – generated by release of glutamate on to postsynaptic neurons. The depolarization generated by AMPA receptors is necessary for effective activation of NMDA receptors. Consequently, AMPA receptor antagonists block most excitatory synaptic activity. Non-NMDA receptors typically pass sodium current, but certain subunit combinations, such as a relative deficit of GluR2, GluR5, or GluR6, endow the receptor with increased calcium permeability, a finding that has implications for development, as well as for epilepsy (see Development of Neurotransmitters, Receptors, and Transporters below) [Rakhade and Jensen, 2009; Santos et al., 2009].
Metabotropic glutamate receptors represent a large, heterogeneous family of G-protein-coupled receptors that subsequently activate various transduction pathways – phosphoinositide hydrolysis and activation of adenylate cyclase and phospholipases C and D. Metabotropic receptors are important modulators of voltage-dependent potassium and calcium channels, nonselective cation currents, and ligand-gated receptors (i.e., GABA and glutamate receptors), and they can regulate glutamate release. Different metabotropic glutamate receptor subtypes are specific for different intracellular processes. Although ubiquitous within the central nervous system, subtypes of metabotropic receptors appear to be differentially localized. Metabotropic glutamate receptors have been implicated in a wide variety of normal neurologic processes (e.g., long-term potentiation) [Anwyl, 2009] and disease states (e.g., epilepsy) [Ure et al., 2006].
Abnormal Neuronal Firing
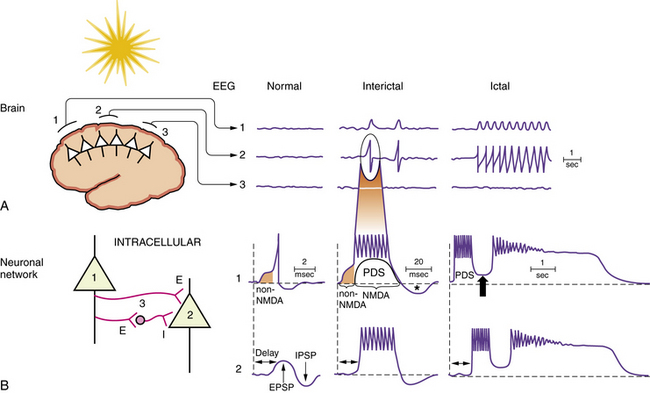
Specific pathophysiological mechanisms mediate each stage of seizure evolution, including transitions from a normal neuronal firing pattern to interictal epileptiform bursts, from interictal firing to seizure activity, and from the seizure to the postictal state [Stafstrom, 2004; Lado and Moshe, 2008]. Figure 51-4 depicts EEG and intracellular changes that can be seen in normal, interictal, and ictal states. In the normal situation, action potentials are generated in neuron 1 when the membrane potential reaches threshold for firing. These discharges may influence the activity of an adjacent neuron (neuron 2) synaptically, resulting in an excitatory postsynaptic potential. An adjacent interneuron (neuron 3, which is inhibitory) may also be activated by a discharge from neuron 1 after a brief delay, giving rise to an inhibitory postsynaptic potential. The activity recorded in neuron 2 reflects the temporal and spatial summation of excitatory and inhibitory postsynaptic potentials.
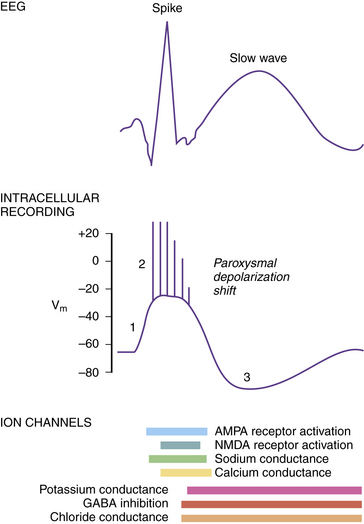
Paroxysmal Depolarization Shift
The intracellular correlate of the focal interictal epileptiform discharge on the EEG is known as the paroxysmal depolarization shift (PDS) [Gorji and Speckmann, 2009]. Initially, there is a rapid shift in the membrane potential in a depolarizing direction, followed by a burst of repetitive action potentials lasting several hundred milliseconds (Figure 51-5). The initial depolarization is mediated by AMPA receptors, whereas the sustained depolarization is a consequence of NMDA receptor activation. Afterward, the PDS terminates with a repolarization phase, primarily a consequence of inhibitory potassium and chloride conductances carried by voltage-gated potassium channels and GABA receptors. The prolonged period of hyperpolarization after the PDS is mediated by inhibitory conductances and constitutes a refractory period. PDS activity in several adjacent neurons would be expected to facilitate synchronous firing [LeDuigou et al., 2009].
< div class='tao-gold-member'>

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


