EPIDEMIOLOGY
An estimated 650,000 AIS occur each year in the United States alone. The most significant individual risk factors for ischemic stroke include hypertension, diabetes mellitus, cigarette smoking, poor nutrition, physical inactivity, cardiac dysrhythmia, alcohol consumption, and obesity. In-hospital mortality after AIS is comparatively rare, occurring in approximately 5% of cases, and is associated with increasing age, stroke severity, history of atrial fibrillation, previous stroke, carotid stenosis, diabetes mellitus, and history of coronary artery disease. Functional disability and morbidity from post-stroke complications occur much more commonly, affecting the majority of patients. As a result, only half of AIS patients are discharged home, whereas the remainder requires further care in skilled nursing facilities, rehabilitation, or palliative care centers.
PATHOBIOLOGY
Despite making up only 2% of total body weight, the brain consumes 20% of the body’s total energy and relies on a constant supply of glucose and oxygen to maintain function and structural integrity. Each minute, nearly 1 L of blood is supplied to the brain through the carotid arteries and, to a lesser extent, the vertebrobasilar system. To ensure a constant cerebral blood flow (CBF) across a range of blood pressures and metabolic states, cerebral vessels constrict or dilate in response to changes in the concentration of carbon dioxide, oxygen, and vessel wall shear stress related to velocity of blood flow. The primary mediator of vasodilation is nitric oxide, which is released by endothelial cells and acts to relax the circumferential smooth muscle cells in the tunica media.
CEREBRAL BLOOD FLOW REDUCTION
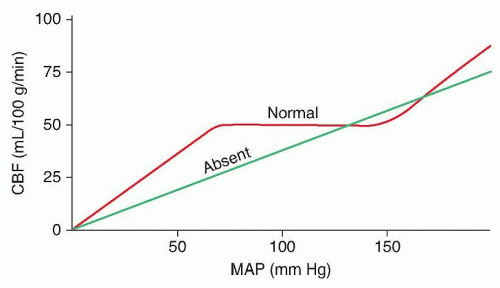
When cerebral perfusion pressure falls below approximately 60 mm Hg due to either systemic hypotension or local arterial blockage, autoregulatory vasodilation cannot compensate leading to decreased CBF from its normal range of 50 to 60 mL/100g/min (
Fig. 35.1). When CBF falls below 30 mL/100g/min, regional exchange of nutrients and oxygen fails within the neurovascular unit (endothelial cell, surrounding pericytes, basement membrane, perivascular astrocytes, and adjoining neurons) resulting in pathologic depolarization of neurons and glia and suppression of the normal electrical activity as evidenced by slowing and attenuation of the electroencephalographic background (
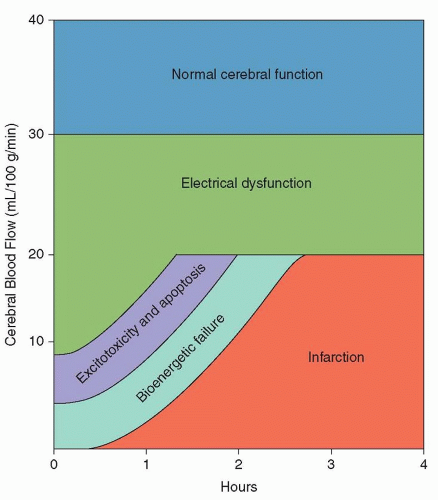
Fig. 35.2). Sustained severe hypoperfusion, with CBF levels falling below 20 mL/100g/min, triggers a cascade of ischemic injury that can lead to irreversible neuronal necrosis and cerebral infarction.
INFARCT CORE AND PENUMBRA
With decreases in CBF below 20 mL/110g/min, assuming a normal rate of neuronal metabolism, brain tissue progresses from a state of reversible electrical dysfunction to a “penumbral” state of excitotoxic damage and programmed cell death (apoptosis). Penumbra is an ancient Latin term that refers to the shadow that advances across the land in advance of the complete darkness of a solar eclipse. In modern stroke therapy, penumbral tissue is brain tissue in the shadow of death.
Failure of the sodium/potassium transmembrane adenosine triphosphate (ATP) pump occurs first, causing neurons and glia
to depolarize. Widespread depolarization causes uncontrolled release of excitatory neurotransmitters such as glutamate, which in turn leads to an intracellular influx of calcium and sodium. Once the phase of excitotoxicity and apoptosis has set in, a phase of bioenergetic failure occurs with ionic pump dysfunction and loss of normal transmembrane sodium and calcium gradients.
In the infarct core, where the bioenergetic failure is greatest, these massive electrolyte shifts cause water to passively follow its osmotic gradient resulting in cellular swelling and eventual lysis. The resultant edema, termed cytotoxic edema as it results from intracellular swelling, manifests within minutes of the initial insult and can be detected with magnetic resonance imaging (MRI) as hyperintensity on diffusion-weighted imaging (DWI) sequences.
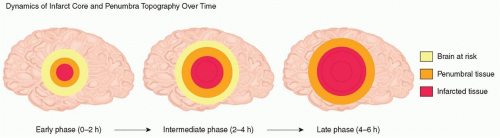
The core of the infarct is surrounded by an area of relative hypoperfusion, the ischemic penumbra, in which CBF is sufficient to temporarily support cell survival but insufficient to maintain normal cellular function indefinitely (
Fig. 35.3). Penumbral tissue may be salvageable if adequate perfusion to the territory can be restored in a timely fashion, which is the basis for all AIS reperfusion therapies. Penumbral tissue, however, is inherently unstable and commonly will progress to completed infarction over time, making
time to reperfusion the most important therapeutic factor for improving neurologic outcome and preventing mortality after AIS.
COMPLETED INFARCTION
It is estimated from human and experimental studies that the time from vessel occlusion to fully completed infarction in most cases ranges from 3 to 6 hours depending on the quality of collateral flow. Once the tissue becomes necrotic and infarcted, it is easily visible on both computed tomography (CT) and fluid-attenuated inversion recovery (FLAIR) MRI images. CBF is maintained at near normal levels up to the margin of a completed infarct where it rapidly drops to undetectable levels as long as the affected vessel remains occluded. Over the next several days, the infarct progressively swells. Ischemic cells that progress to frank tissue infarction release various proinflammatory cytokines and proteases that disrupt the architecture and function of the neurovascular unit, increasing permeability of the blood-brain barrier. Apoptosis is triggered through the function of various inflammatory processes, including the receptor/ligand combination of the CD95 receptor and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and mitochondrial membrane release of cytochrome c oxidase, both of which lead to activation of the intrinsic programmed cell death cascade and delayed cell death. This delayed neurologic injury could in theory be prevented with neuroprotective agents, although none has proven to be of use in clinical trials.
The parenchymal inflammatory response results from upregulation of endothelial cell leukocyte adhesion receptors that cause
leukocyte adhesion and transmigration into the affected tissue and is responsible for the prolonged process of tissue inflammation and remodeling after an infarct is completed, as the swelling resolves and the involved regions convert to gliotic scar tissue.
ETIOLOGY OF ISCHEMIC STROKE
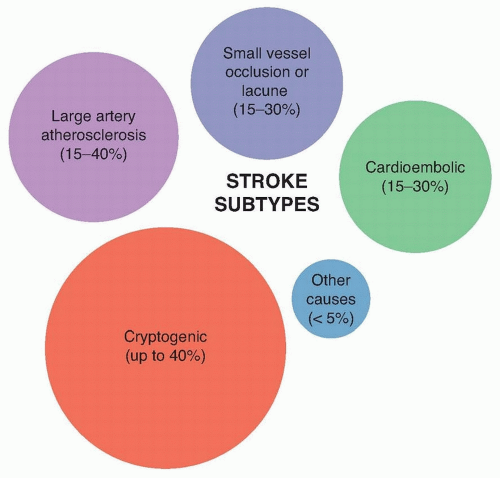
The microscopic process of acute cerebral infarction is preceded by macrovascular occlusion of the territory in question. Accurate classification of AIS etiology is important to understand post-stroke prognosis, quantify the risk of recurrence, and guide strategies for secondary stroke prevention. Although imperfect, the Trial of Organon in Acute Stroke Treatment (TOAST) classification criteria is the most widely accepted ischemic stroke classification system and includes the following five subtypes: large artery atherosclerosis (15% to 40%), cardioembolic (15% to 30%), small-vessel occlusion or “lacunar” (15% to 30%), cryptogenic (up to 40%), and “other” cause (<5%) (
Fig. 35.4).
LARGE-ARTERY ATHEROSCLEROSIS
Atherosclerotic plaque may develop at any point along the extracranial or intracranial arterial tree but is usually seen at vessel-branch points where turbulent flow occurs, most commonly at the common carotid bifurcation, vertebral artery origin, vertebrobasilar junction, or the origin of the middle or anterior cerebral arteries. Atherosclerotic plaque causes ischemic stroke by one of two mechanisms: hypoperfusion across a region of critical stenosis or, more commonly, plaque rupture leading to thrombus formation and subsequent distal embolization of thrombus or plaque fragments.
Artery-to-artery embolism occurs when fragments of atherosclerotic plaque or fresh thrombus formed on top of the plaque embolize to occlude a distal vessel. Plaques with a lipid-rich necrotic core and thin fibrous cap pose a risk of spontaneous rupture and activation of the coagulation cascade leading to superimposed thrombus formation. Biomechanical forces induced by turbulent blood flow along this irregular portion of vessel can act to destabilize the cholesterol plaque or dislodge a superimposed thrombus. When this happens, the thrombus may rapidly dissolve causing the clinical symptoms to be fleeting or absent, or it may persistently occlude a distal vessel leading to lasting symptoms of AIS.
Clinically, ischemia from large-artery atherosclerosis is difficult to distinguish from stroke originating from the heart (cardioembolic stroke). Favoring the former are repeated transient ischemic attacks or completed infarcts within the same vascular territory, the best example of which is repeated episodes of painless transient monocular blindness related to ipsilateral carotid artery atherosclerosis. This condition, known as amaurosis fugax, results from cholesterol embolism from the carotid plaque causing occlusion of the central retinal artery or one of its branches. When the lumen of the carotid artery is nearly occluded, transient attacks can also be due to retinal hypoperfusion, clinically manifesting as vision loss with exposure to bright light. Similarly, repeated episodes of vertigo, diplopia, hemiparesis, hemianesthesia, gait unsteadiness, and even loss of consciousness can be a sign of high-grade vertebral or basilar artery stenosis.
CARDIOEMBOLIC STROKE
Cardioembolic stroke includes all strokes caused by a cardiac source of thromboembolism. Most commonly, cardioembolism occurs from thrombus formed in the left atrial appendage of the heart in patients with atrial fibrillation, but cardioembolism can also result from a diseased native or prosthetic heart valve or ventricular wall thrombus resulting from recent myocardial infarction or severe dilated cardiomyopathy. Cardioembolic strokes can also result from neoplastic causes, such as atrial myxoma or marantic endocarditis, or infectious causes such as infectious endocarditis. Radiologically, scattered strokes at the gray-white matter junction that occur in both hemispheres, or in both the anterior and posterior circulation, are most commonly cardioembolic in origin. However, strokes that appear in association with proximal large-vessel
occlusions or that appear most consistent with small-vessel infarction can also be seen after cardioembolism.
SMALL-VESSEL INFARCTION
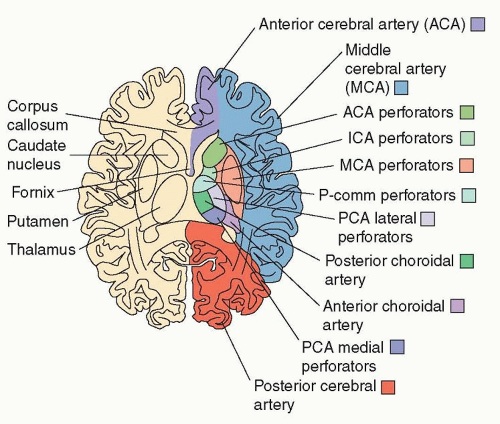
Lacunar strokes from small-vessel occlusion are clinically defined by five general subtypes: (1) pure motor hemiparesis, (2) pure sensory syndrome, (3) mixed sensorimotor syndrome, (4) ataxic hemiparesis, and (5) clumsy-hand dysarthria, although various other manifestations can occur depending on the territory of the vessel involved. These strokes result from blockage of small perforating arteries arising from the middle cerebral artery (lenticulostriate), the posterior cerebral or posterior communicating arteries (tuberothalamic, paramedian, posterior choroidal, inferolateral), and basilar artery (pontine perforators). These vessels are prone to segmental arterial wall disorganization and fibrosis, a process known as lipohyalinosis, and microatheroma formation. Over time, the arterial wall thickens and the vessel lumen is compromised causing a region of acute ischemia that is usually smaller than 1.5 cm in diameter. Hypertension is the greatest risk factor, although diabetes, age, and smoking may contribute as well. Lacunar infarction occurs most commonly in the basal ganglia, thalamus, internal capsule, corona radiata, and pons. Imaging helps distinguish lacunar stroke from other types of stroke by their subcortical location and relatively small size.
UNDETERMINED CAUSE OF STROKE
The etiology of up to 40% of strokes remains cryptogenic, or undetermined, even after extensive laboratory and radiologic investigation. However, many of these patients may in fact have undiagnosed atrial fibrillation with studies showing that 10% to 20% of “cryptogenic” stroke patients are found to have occult atrial fibrillation after prolonged cardiac monitoring. Although patent foramen ovale (PFO) and atrial septal defect are also associated with cryptogenic stroke in younger patients, PFO closure is not yet of proven efficacy for the prevention of stroke recurrence.
OTHER CAUSES OF STROKE
The “other” causes of stroke category makes up less than 5% of AIS, and it is defined by a specific disease process that is shown to have a temporal relationship or association to the stroke. In addition, diagnostic investigation should not reveal any other more-common mechanism. Examples include stroke due to hematological disorders (e.g., hypercoagulability, sickle cell anemia), infectious or inflammatory disease, intrinsic disease of the arterial wall (i.e., vasculopathy), vessel dissection, migraine-associated infarction, genetic disorders, or iatrogenic causes.
CLINICAL MANIFESTATIONS
Ischemic strokes can be defined clinically by the nature of the associated neurologic deficit with signs and symptoms fitting into defined syndromes representing specific vascular territories (
Fig. 35.5). Knowledge of the following major stroke syndromes can oftentimes accurately predict the territory of infarction and specific vessel involved (
Table 35.1). Exceptions arise when there is underlying preexisting CNS injury, presence of collateral circulation, or variations in the vascular or brain anatomy that can produce atypical or unexpected clinical presentations.
MIDDLE CEREBRAL ARTERY INFARCTION
Infarction of the middle cerebral artery (MCA) produces clinical syndromes that depend on the extent of the infarct and the side and level of vessel occlusion. Proximal, or branch MCA occlusion, can affect both the deep and the cortical hemispheric structures or deep structures only when there is robust collateral vascularization supplying the cortex. Isolated ischemia of the deep cerebral structures—mainly the internal capsule, basal ganglia, and corona radiata—occurs due to blockage of the lenticulostriate artery origins causing contralateral hemiparesis and possibly hemisensory loss that often is indistinguishable from a lacunar syndrome. Larger deep infarcts (“striatocapsular infarction”) that affect cortical connections to the thalamus and optic radiations can cause the cortical syndromes of hemineglect, aphasia, and homonymous hemianopia or quadrantanopia.
Cortical ischemia in the MCA territory is encountered in isolation when vessel occlusion is localized to the superior or inferior MCA divisions (the number and location of these branch arteries can vary). Damage to either hemispheric cortex can cause contralateral limb weakness, contralateral sensory loss, astereognosia (inability to identify objects using touch), agraphesthesia (inability to recognize writing traced on the skin), diminished two-point discrimination, contralateral visual field deficits, and gaze deviation towards the damaged side. Ideational apraxia (inability to understand the purpose of an object or manipulate it) follows left-sided or bilateral hemispheric damage.
Syndromes resulting from a dominant (usually left) hemispheric lesion include aphasia (language dysfunction), bilateral ideomotor apraxia (inability to perform learned motor tasks), and buccolingual apraxia (inability to voluntarily direct mouth and tongue movements). The type and severity of aphasia depends on the location and size of the infarct. A superior MCA branch occlusion will affect the anterior perisylvian cortex and specifically the inferior frontal gyrus to produce Broca (expressive) aphasia that is characterized by decreased or absent word fluency and inability to write but with preserved comprehension. When aphasia is expressive or global, hemiparesis is also usually present and severe. Inferior branch MCA occlusion, affecting the posterior perisylvian cortex and specifically the superior temporal gyrus and temporoparietal junction, produces Wernicke (receptive) aphasia, which is characterized by preserved fluency and prosody but incomprehensible content, semantic and phonemic paraphasic errors, impaired comprehension, and inability to read, name, or repeat.
Receptive aphasia is often associated with a homonymous hemianopia or quadrantanopia, but hemiparesis can be mild or even absent. Gerstmann syndrome (left-right confusion, finger agnosia, acalculia, and agraphia) follows infarction of the dominant angular gyrus.
Infarction of the nondominant (usually right) parietal lobe produces contralateral hemispatial neglect or an inattention to sensory inputs or motor outputs to the contralateral half of the environment or body. Specific findings include visual neglect (that can involve the entire contralateral visual field or be specific to various objects), anosognosia (lack of awareness of neurologic deficits), asomatognosia (inability to recognize one’s limb on the affected side), and allochiria (spatial transposition resulting in the patient attending to the ipsilateral hemispace when presented with a stimulus on the neglected contralateral side). Constructional apraxia (inability to assemble or draw objects) may result from damage to either hemisphere but is more obvious in nondominant hemispheric lesions. Although speech content or the propositional aspect of speech is preserved, a nondominant hemispheric lesion may cause deficits in the pragmatics, or sociolinguistic aspects of speech, involving tone and inferred intent.
ANTERIOR CEREBRAL ARTERY INFARCTION
Anterior cerebral artery (ACA) infarction most commonly causes contralateral leg hemiparesis and/or hemisensory loss, although many other features may also be seen. Similar to watershed infarcts, ACA territory infarcts can cause transcortical aphasia of the motor type. Motor neglect, or a disinclination to use the contralateral limb despite the absence of weakness, is often encountered and is due to dysfunction of the prefrontal supplementary motor cortex. Bilateral ACA infarction often results in executive dysfunction with motor inertia and abulia (paucity of spontaneous behaviors) or even akinetic mutism (awake unresponsiveness). Gait apraxia, although typical of bilateral lesions, can also result from unilateral strokes. Alien hand syndrome (unwilled motor activity) or hemiballism of the contralateral arm can result from injury to the anterior corpus callosum or medial frontal cortex. Utilization and imitation behavior are neurobehavioral syndromes that can occur with unilateral prefrontal infarcts. With utilization behavior, the patient automatically and impulsively grabs objects that appear within visual field and are within reach. Imitation behavior is automatic and involuntary imitation of the examiner’s movements.
Occlusion of the recurrent artery of Heubner, a large perforating artery arising from the proximal ACA, leads to infarction of the head of the caudate, anterior internal capsule, and anterior putamen. Common clinical features include dysarthria, motor perseveration, contralateral hemiparesis, abulia, and incontinence.
INTERNAL CAROTID ARTERY INFARCTION
Acute carotid occlusion causes acute hemispheric hypoperfusion that, in the absence of a patent circle of Willis, can manifest as seizure, contralateral limb shaking, or holohemispheric infarction of both the ACA and MCA territories. Occasionally, and especially in the presence of a robust circle of Willis, occlusion of the internal carotid artery (ICA) spares the ACA and produces a clinical scenario indistinguishable from branch MCA infarction. If collateral
flow across the posterior communicating artery, anterior communicating artery, or pial arteries is adequate, the occlusion can be clinically silent or only affect smaller susceptible areas. By contrast, ICA occlusion with poor collateral or a distal ICA “T occlusion” can lead to catastrophic holohemispheric infarction with sparing of only the posterior cerebral artery (PCA) territory (thalamus and medial temporal and occipital lobes).
BORDER ZONE INFARCTION
Border zone (or watershed) territories occur at the boundaries between the anterior, middle, and posterior cerebral arteries and are susceptible to ischemia in acute carotid occlusion or global hypoperfusion.
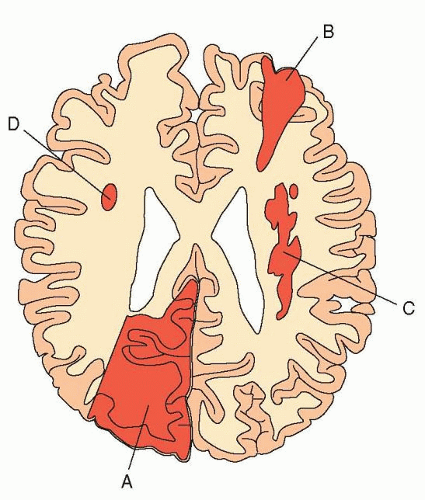
Superficial border zone infarction occurs high along the convexity of the corona radiata between the deep and cortical structures anteriorly between the ACA and MCA territories or posteriorly between the MCA and PCA territories (
Fig. 35.6). This pattern of infarction occurs when collateral flow is able to supply the majority of the involved territories but is unable to “meet” between territories. Resulting symptoms form a combination of weakness, hemisensory loss, and cortical signs. Transcortical aphasia of the expressive or receptive type with relatively preserved ability to repeat is also common. ACA-MCA watershed infarcts primarily cause arm weakness because the affected region corresponds anatomically with neurons in the motor strip that innervate the upper extremities. When bilateral, as occurs with cardiac arrest, ACA-MCA watershed infarctions result in
man-in-the-barrel syndrome characterized by proximal bilateral arm and leg weakness with relative preservation of facial and distal extremity strength.
Internal border zone infarction (see
Fig. 35.6) can occur with total ICA occlusion or with large MCA branch occlusions. Noncollateralized deep penetrating arteries that descend from the cortex to the lateral wall of the ventricles represent the most hemodynamically vulnerable “distal field” in this scenario. The result is a pattern of multiple small deep infarcts involving the deep white matter of the corona radiata and centrum semiovale.
ANTERIOR CHOROIDAL ARTERY INFARCTION
The anterior choroidal artery is the last branch of the ICA originating just proximal to the ACA-MCA bifurcation. It traverses posteriorly to supply a number of structures, including the internal capsule, lateral thalamus, and the lateral geniculate body or optic radiations. Thus, occlusion of the anterior choroidal artery results in the classic syndrome of contralateral hemiparesis, hemianesthesia, and homonymous hemianopia. Occasionally, cortical symptoms such as aphasia or hemineglect can result from disruption of thalamocortical white matter tracts.
POSTERIOR CEREBRAL ARTERY
Occlusion of the PCA will cause infarction of the occipital and inferior temporal lobes. The usual presentation is sudden onset of a contralateral homonymous hemianopia. Bilateral occipital lobe lesions can cause Anton syndrome (cortical blindness unrecognized by the patient). Charles Bonnet syndrome, or release visual hallucinations, can also be seen in cortical blindness from bilateral occipital lobe injury. Balint syndrome is seen in bilateral parietooccipital junction infarcts and is characterized by simultanagnosia (inability to perceive parts of an object as a whole), oculomotor apraxia (incoordination of gaze), and optic ataxia (poor hand coordination using visual guidance). Hemineglect, although uncommon with occipital lesions, can be seen when the injury extends to the parietal lobe. Left PCA infarcts that involve either the deep white matter of the temporoparietal junction or the splenium of the corpus callosum may cause alexia without agraphia, a type of disconnection syndrome between the nondominant occipital cortex and the language processing area in the dominant parietotemporal cortex. Inferomedial temporal lobe infarction may cause agitated delirium or global amnesia.
Proximal PCA infarction affecting the thalamic perforating arteries will lead to infarction of the thalamus or midbrain. If the ventral posterior nucleus of the thalamus is preferentially affected, the patient will have contralateral hemianesthesia. Hyperpathia may subsequently develop in a condition called the Dejerine-Roussy or thalamic pain syndrome. As the thalamus serves as a relay to the cortex, thalamic infarction can also cause a variety of atypical symptoms such as ataxia, aphasia (dominant), hemineglect (nondominant), visual field deficits, memory loss, or behavioral disturbances.
Lethargy or coma can result if the reticular activating system in the midbrain or thalami is affected and will often be accompanied by a combination of oculomotor abnormalities. Alterations in consciousness are especially common in the presence of an anatomically variant artery, termed the artery of Percheron, which supplies the bilateral medial thalami and rostral midbrain from a single vessel originating on a unilateral PCA. Midbrain lesions, when posterior, cause Parinaud syndrome characterized by sustained conjugate downgaze, upgaze paralysis, convergence or retraction nystagmus, lid retraction or Collier sign, and pupillary light-near dissociation.
VERTEBROBASILAR TERRITORY INFARCTION
The basilar artery is the main conduit to supply the PCA arteries and basilar thrombosis can manifest with a combination of medullary, pontine, midbrain, and posterior cortical signs. Basilar thrombosis
leading to infarction of the anterior pons bilaterally causes “locked-in” syndrome: complete bilateral face, arm, and leg paralysis with isolated preservation of vertical eye movements and preservation of consciousness (see
Chapter 18). Infarcts resulting from small-vessel thrombosis of the pontine perforating arteries cause a combination of cranial nerve dysfunction, long tract signs including weakness, reflexive posturing or hyperreflexia, sensory loss, and/or cerebellar findings. Small anterior pontine strokes can also produce lacunar syndromes without cranial nerve deficits that are clinically indistinguishable from anterior circulation lacunes. Oculomotor or gaze palsies, including internuclear ophthalmoplegia and skew deviation, or nystagmus, are also common. “Top of the basilar” syndrome refers to occlusion of bilateral distal basilar and proximal PCA thalamic and midbrain penetrating arteries from embolism. The result is a pattern of bilateral small-vessel infarcts involving the midbrain and thalami with asymmetric vertical gaze abnormalities and skew deviation, changes in level of consciousness, and variable bilateral motor and sensory deficits.
Infarction of the lateral medulla occurs when the posterior inferior cerebellar artery or a branch of the vertebral artery is affected. What results is a lateral medullary or
Wallenberg syndrome, which is characterized by vertigo, ipsilateral ataxia, loss of pain and temperature sense in the ipsilateral face and the contralateral arm and leg, dysphagia, and ipsilateral oculosympathetic syndrome (Horner syndrome). Other brain stem infarct syndromes are described in
Table 35.2.