and Kelly Del Tredici1
(1)
Zentrum f. Biomed. Forschung AG Klinische Neuroanatomie/Abteilung Neurologie, Universität Ulm, Ulm, Germany
8.1 The Amyloid Precursor Protein and the Abnormal Protein Aβ
A clear indicator for the end of the unusually protracted initial phase of the AD-associated pathological process is the abrupt appearance of an additional protein that appears in soluble form in the ISF: the small, i.e., 38–43, but mostly 40 or 42, amino acid-containing hydrophobic amyloid-β (Aβ) protein that at first is diffusely distributed in a monomeric state in a few circumscribed regions of the ISF but then rapidly forms insoluble aggregations, most of which are plaque-like entities. These Aβ-plaques develop with such consistency in the course of AD that they constitute one of its hallmark lesions (Masters and Selkoe 2012).
The pathological Aβ peptide is generated by abnormal proteolytic processing of a physiological constituent of the nerve cell membrane, the amyloid precursor protein (APP) (Beyreuther and Masters 1991; Mattson 2004; Rajendran and Annaert 2012). APP is an integral membrane glycoprotein that presumably functions as a receptor. In addition, APP has been ascribed neurotropic and neuroprotective properties (Selkoe 1994; Selkoe et al. 2012).
For the most part, APP is degraded without a trace by a process that does not permit Aβ production (Fig. 8.1a). During this process, α-secretase splices the APP and generates a soluble molecule (APPsα) that is released into the ISF. The remaining membrane-bound fragment (C83) is spliced by γ-secretase, and an additional non aggregation-prone fragment (P3) is released into the ISF, whereas the leftover APP C-terminal domain (AICD) remains in the neuronal cytoplasm (Fig. 8.1a) (Haass et al. 2012).
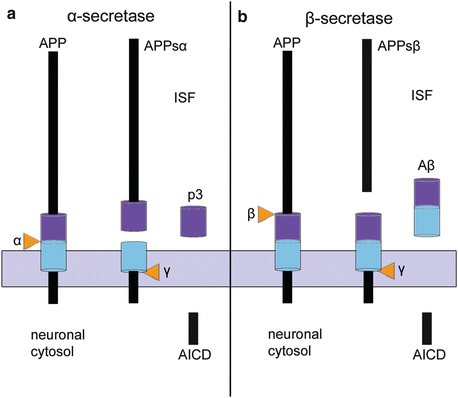
Fig. 8.1
Two processing pathways for the amyloid precursor protein. (a) The normal pathway utilizing α-secretase prevents the formation of Aβ and only produces p3 while, in (b), processing with β-secretase leads to the production of Aβ. The pathway displayed in (b) only occurs in a few vulnerable types of nerve cells. Diagrams adapted and reproduced with permission from C Haass et al., Trafficking and proteolytic processing of APP. Cold Spring Harb Perspect Med 2012; 2:a006270. Abbreviations: AICD APP intracellular C-terminal domain, APP amyloid precursor protein, APPsα soluble α-remnant of APP, APPsβ soluble β-remnant of APP, ISF interstitial fluid
Aβ comes into existence only under pathological conditions and originates via an abnormal degradation pathway. First, a long and soluble fragment (APPsβ) is cleaved from APP by a β-secretase (Fig. 8.1b). The membrane-anchored fragment (C99) is subject to further clearance via γ-secretase, and the result is the release of Aβ into the ISF, whereas the leftover AICD remains in the neuroplasm. This sequential cleavage by β- and γ-secretases is thought to occur in the weakly acidic environment of recycling endosomes (Haass et al. 2012).
Because these steps all take place within nerve cells, the interpretation of experimental results emerging chiefly from non-polarized cells is problematic. Nonetheless, polarized cell models show that the enzymes α- und β-secretase can be distributed very differently, so that it is plausible that Aβ production by means of β-secretase can occur only at specific and predetermined sites and only in select types of nerve cells. By contrast, as anticipated, the degradation process via γ-secretase takes place at all APP-cleavage sites (Haass et al. 2012). Moreover, it is known that APP undergoes vesicular anterograde transport within axons. Thus, terminal axons and preferably presynaptic varicosities could turn out to represent the major secretion sites of Aβ (Lazarov et al. 2005).
8.2 Sources and Secretion of Aβ
Previous findings have shown that the Aβ peptide does not enter the ISF from the serum, from the vasculature, ependymal organs, or the choroid plexus. In addition, neither astrocytes, oligodendrocytes, nor microglia cells generate Aβ (Beyreuther and Masters 1991; Fiala 2007). The current consensus is that nerve cells are the sole sources of Aβ. It is very unlikely, however, that essentially all types of nerve cells within the nervous system produce Aβ because Aβ plaques are found only in portions of the CNS and not in the ENS or PNS. In addition, Aβ deposits do not occur with the same frequency or severity in all regions of the CNS (see also Sect. 8.5). Thus, similar to tau aggregation, Aβ deposition occurs in the CNS only at specific sites and according to a consistent developmental distribution pattern (Braak and Braak 1991a; Thal et al. 2002).
Generally, Aβ deposits in AD rarely develop in the white substance; instead, they mainly occur in the gray matter, including nerve cell somata and cellular processes of nerve cells (Figs. 8.2–8.4). In the gray matter, it is possible to distinguish regions with high densities of Aβ plaques, e.g., the anterior olfactory nucleus and olfactory bulb (Kovács et al. 1999; Attems and Jellinger 2006), the entire neo- and allocortex (Thal et al. 2002), claustrum, striatum (Braak and Braak 1990; Beach et al. 2012b), thalamus, mesencephalic tectum, red nucleus, cerebellar cortex (Braak et al. 1989b), and specific locations of the lower brainstem, from sites where Aβ plaques are sparse, e.g., the multiform layer of the neocortex, the lateral geniculate body of the thalamus, substantia nigra, and the precerebellar nuclei in the brainstem, among others. Aβ plaques are absent in both segments of the pallidum as well as in the hypothalamic lateral tuberal and lateral mamillary nuclei. This pattern of Aβ plaques occurs with little inter-individual variability and is the major reason for surmising that not all types of nerve cells of the CNS can produce Aβ.
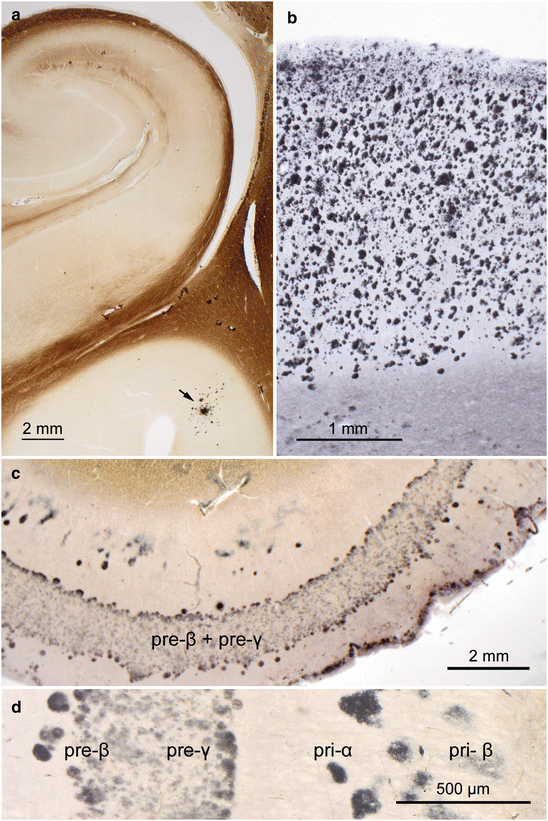
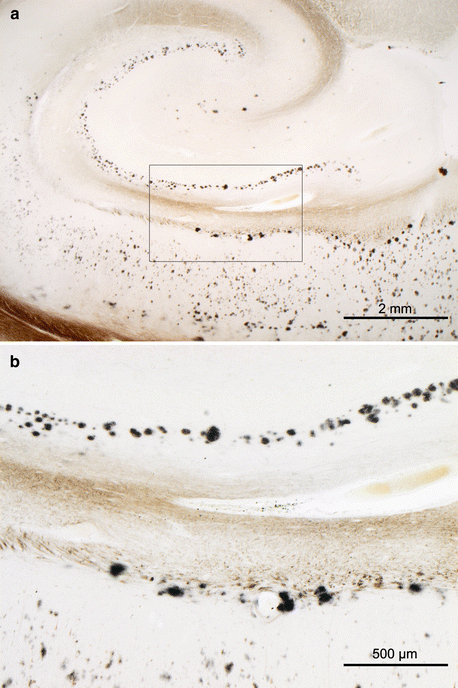
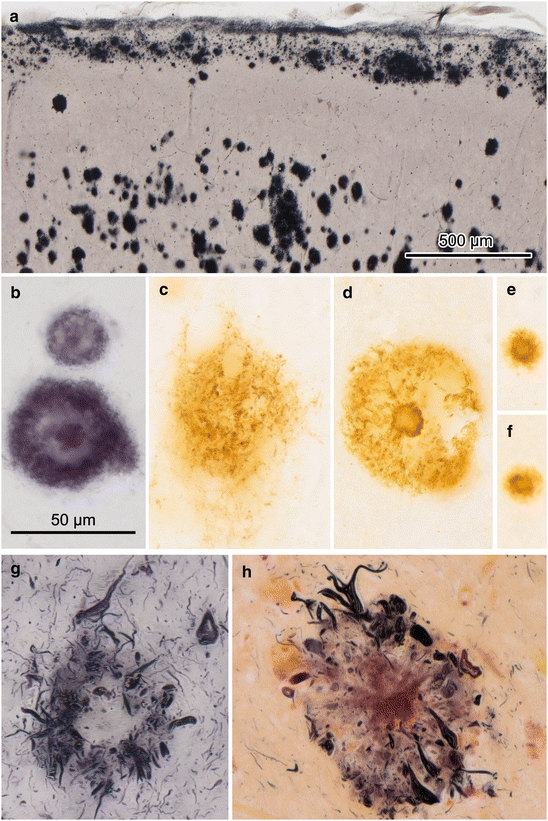
Fig. 8.2
Aβ plaques in 100 μm sections processed with the Campbell-Switzer silver-pyridine technique. (a) Phase 1: Initially, isolated plaques develop in the basal temporal neocortex (arrow) in the absence of plaques in the hippocampal formation (42-year-old male). (b) Phase 5: Maximal plaque density in the temporal neocortex of an 82-year-old demented male with AD (NFT stage V). (c) Band-like plaque formation in layers pre-β and pre-γ of the entorhinal region of in 87-year-old male AD patient (NFT stage V), seen in greater detail in (d). Other amyloid precipitations, such as those occurring in prion diseases (spongiform encephalopathies), remain unstained. Fully developed silver-stained sections demonstrate a non-specific co-staining of axons. This readily and reliably applicable silver technique also distinctly demonstrates neuromelanin granules and Lewy bodies/neurites in Lewy body disease (PD) as well as argyrophilic oligodendrocytes associated with multisystem atrophy (MSA). See also the Technical addendum in Chap. 11.
Fig. 8.3
Aβ plaques in 100 μm sections (Campbell-Switzer silver-pyridine method). (a) Phase 3: Aβ deposits develop in the hippocampal formation of a 67-year-old female. Note the densely packed row of plaques along the course of the perforant pathway not only in CA 1 but also in the molecular layer of the dentate fascia. (b) Higher magnification of the framed area in (a)
Fig. 8.4
Different forms of Aβ plaques in 100 μm sections. (a) Band-like deposits of Aβ directly subjacent the layer of surface astrocytic endfeet. Deeper portions of the molecular layer harbor densely packed globular plaques that frequently become confluent (female, 96 years of age, NFT stage VI). (b, d) Examples of cored plaques in an 84-year-old male (b, NFT stage V, Campbell-Switzer) and in a 60-year-old male (d, stage VI, 4G8 immunoreaction). (c) Diffuse plaques often show ill-defined surfaces (same stage VI case as in d, 4G8 immunoreaction), whereas cored plaques (d) mostly have clear-cut outlines. (e, f) Burned out plaques are much smaller and generally have a core (same individual as in d, 4G8 immunoreaction). (g, h) Examples of NPs in a 74-year-old male (g) and in a 60-year-old male (h). Gallyas silver-iodide impregnations stain a network of argyrophilic neuronal processes in peripheral portions of amyloid deposits. The amyloid core is unstained in (g) and differently stained in a violet shade in (h). Scale bar in (b) applies also to (c–h)
As pointed out earlier (Sect. 2.2), CNS neurons can have a long or a short axon (Fig. 2.1e–g). The characteristic Aβ distribution pattern associated with the AD process makes it improbable that nerve cells with a short axon contribute to Aβ production because, were this to be true, one should see precipitations of Aβ in the immediate vicinity of these cells; but that does not happen. Therefore, the number of CNS nerve cell populations that produce Aβ cannot be, by process of elimination, very large. Of course, the question arises whether all projection neurons with a long axon can generate Aβ under normal conditions. If so, an ongoing Aβ production should be detectable throughout the lifetimes of all individuals irrespective of their cognitive status. However, inasmuch as there is no evidence for such a generalized process, it is clear that Aβ production is integral to the AD-associated process. Presumably, the homeostasis of projection neurons that have AD-associated intraneuronal lesions is not unperturbed. For this reason, it is possible that Aβ originates chiefly, or perhaps solely, from CNS projection neurons with tau pathology.
The fact that the somatodendritic domains of involved projection neurons are seldom surrounded by Aβ deposits raises the question at which cellular sites specifically (dendrites, soma, axon, synapses) Aβ is released into the ISF. Given what is already known about the typical plaque distribution pattern (Thal et al. 2002), it can be ruled out that Aβ is released via dendrites or cell bodies. In addition, it can be surmised that Aβ is not given off through most of the axonal membranes (for instance, at the nodes of Ranvier) because the white matter remains nearly devoid of Aβ deposition and only a few plaques are seen to develop near the cortical gray matter. Instead, Aβ deposits are more or less evenly distributed among the somatodendritic domains of nerve cells. Direct contacts with neurons occur only on a random basis and as a result of the high densities of both nerve cells and Aβ deposits (Fig. 8.2b). No direct evidence indicates a potential release of Aβ via the somatodendritic domain. Notably, some sites that harbor cell somata and dendritic processes with neurofibrillary changes, such as the locus coeruleus or layer pre-α of the entorhinal region, remain free of Aβ deposition (Fig. 8.2c). Involved coeruleus neurons have tau-immunoreactive inclusions in both dendrites and axons. However, whereas the axons extend into the cerebral cortex, which is richly supplied with Aβ plaques, the dendrites remain confined to the local neuropil of the brainstem, which contains very few plaques. Therefore, it is unlikely that Aβ is released from dendrites. Moreover, it has been shown that APP is transported along axons (Koo et al. 1990). This finding and the distribution pattern of Aβ plaques in general make it more likely that Aβ is released from presynapses of terminal axons, along which nerve cells normally release their neurotransmitter and/or neuromodulator substances (Stokin and Goldstein 2006; Muresan and Muresan 2008; Harris et al. 2010; Haass et al. 2012; Braak and Del Tredici 2013a).
In the course of the AD process, plaque-like Aβ deposits do not occur in the absence of intraneuronal tau pathology—they develop later than the tau lesions (Table 7.2; Fig. 9.16) (Silverman et al. 1997; Schönheit et al. 2004; Dong et al. 2012; Giacobini and Gold 2013; but see Hardy and Selkoe 2002; Price and Morris 2004; Hardy 2006; Golde et al. 2011; Karran et al. 2011; Mann and Hardy 2013). This means that Aβ deposition begins when specific types of nerve cells, e.g., nerve cells in the brainstem nuclei with diffuse cortical projections, already have undergone cytoskeletal tau changes. The assumption that Aβ is the initial causal event of the AD process is therefore erroneous (compare Fig. 9.16a and b) (Korczyn 2008; Pimplikar 2009; Duyckaerts 2011; Braak and Del Tredici 2013a, b; Jagust et al. 2012; Chételat 2013; Chételat and Fouquet 2013; Perani 2014).
First, primitive (i.e., diffuse) Aβ plaques develop in the basal temporal neocortex (Braak and Braak 1991a) (Fig. 8.2a); in other words, at a time and in a region where pyramidal cells lack AD-associated tau aggregations. If our assumption is correct that Aβ only originates in nerve cells that are already involved in the AD process, then Aβ can only reach the basal temporal neocortex by way of long axons projecting to this part of the neocortex, a condition fulfilled by the axons of the diffusely projecting brainstem nuclei.
The existence of Aβ plaques in the cerebellum (Braak et al. 1989b; Thal et al. 2002) can best be explained by a similar phenomenon, i.e., the release of Aβ via terminal axons belonging to nerve cells with tau pathology, insofar as the various cerebellar neuronal types do not develop abnormal tau inclusions. They are, however, well supplied with a dense axonal network originating from brainstem nuclei, above all the locus coeruleus, where abnormal tau inclusions occur remarkably early.
In this context, it is necessary to reiterate that the terminal segment of the extensively branching axons of diffusely projecting brainstem nuclei develop large numbers of local thickenings with only presynaptic sites (so-called “non-junctional varicosities”) in the absence of postsynaptic counterparts. By means of these varicosities, they release their neurotransmitter and neuromodulator substances (volume transmission) diffusely into the ISF (Agnati et al. 1995; Nieuwenhuys 1999; O’Donnell et al. 2012). It is conceivable that soluble forms of Aβ may likewise be released at non-junctional varicosities directly into the ISF (Braak and Del Tredici 2013a). This interpretation is supported by the existence of Aβ deposits that are found around the smooth muscle layer of vessel walls in the CNS in the form of cerebral amyloid angiopathy (CAA) (Yamada and Naiki 2012) (see Sect. 8.7). Moreover, since axons of the diffusely projecting brainstem nuclei only spread throughout the CNS—a volume transmission mechanism would also account for why Aβ plaque formation remains confined to the CNS and does not develop in the PNS and ENS (for the olfactory mucosa, however, see Arnold et al. 2010).
With the notable exception of Aβ plaques in the striatum the dense network of coeruleus noradrenergic terminals corresponds remarkably well to the topographic distribution pattern of both Aβ plaques and CAA in sporadic AD (Counts and Mufson 2012). It still must be shown whether Aβ is preferentially given off from terminals of coeruleus neurons and whether additional nuclei with diffuse projections also contribute to the production of Aβ plaques, such as the terminals of the upper raphe nuclei, which, in turn, could explain the development of Aβ plaques in the striatum (Braak and Del Tredici 2013a).
The pallidum is an expansive forebrain region that is not reached by ascending projections originating from noradrenergic, serotonergic, or cholinergic non-thalamic nuclei. This fact accounts for the previously mentioned and puzzling finding that both segments of the pallidum belong to the very few regions of the forebrain that do not develop Aβ plaques. Unclear is whether a similar relationship can also be found for the absence of Aβ deposits in selected regions of the hypothalamus (i.e., the lateral tuberal nucleus and lateral mamillary nucleus).
The perforant pathway also deserves mention because it is frequently decorated with Aβ deposits (Fig. 8.3). Projection cells in the external entorhinal cellular layers give rise to this glutamatergic path that terminates in the hippocampal formation (CA 1 and dentate fascia) (Hyman et al. 1988; Braak et al. 1996). The host entorhinal neurons tend to develop intraneuronal tau inclusions early, and Aβ deposits are often found later close to the terminal ramifications of their axons. Perforant pathway fibers contact only a portion of the dendritic tree of CA 1 projection neurons, whereas dendritic segments outside of the pathway are initially free of Aβ deposits. For these reasons, axon terminals of the perforant path may also be capable of releasing Aβ (Fig. 8.3) (Buxbaum et al. 1998; Harris et al. 2010). It is still not known whether axons of the perforant path are endowed with non-junctional varicosities.
In the ISF, the hydrophobic but still soluble Aβ molecules are prone to further aggregation that may be induced by seeding sites. The pathological material ultimately converts into insoluble plaque-like deposits of variable sizes and shapes (Figs. 8.2–8.5) (Thal et al. 2002). Insoluble Aβ precipitations can be visualized using Campbell-Switzer silver-pyridine staining or immunoreactions (Campbell et al. 1987; Braak and Braak 1991b; Montine et al. 2012). The aggregated amyloid fibrils of primitive or cored plaques in the cortex are rich in cross-β sheet structures (Haass et al. 2012; Masters and Selkoe 2012), whereas these components in the non-amyloid Aβ plaques of the striatum and cerebellum are sparse. Because aggregated fibrils possess low bioactivity, we are inclined to see them as posing no immediate danger to adjoining components of the neuropil. From then on, the potentially undesirable side-effects of the Aβ plaques essentially would arise from their capacity to displace other structures. In fact, given the limited dimensions of the extracellular space in the CNS and its importance for the functionality of nerve cells, it is certainly conceivable that such side-effects could occur.
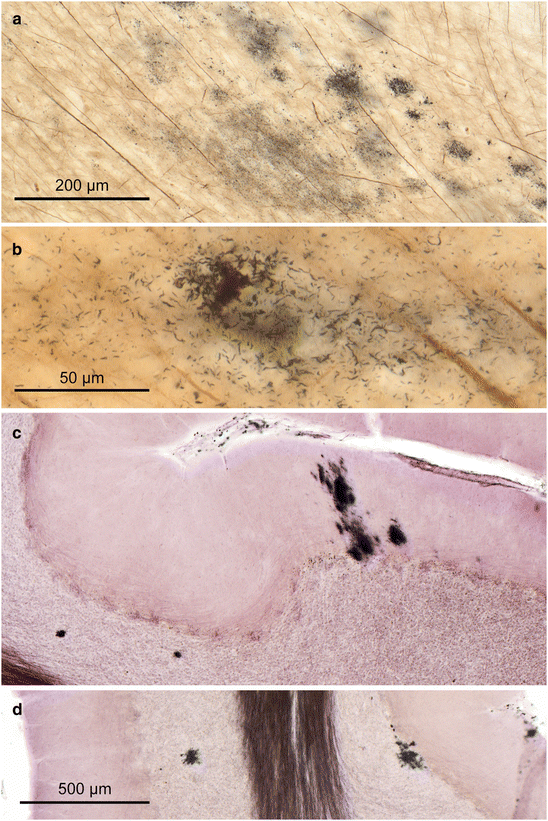
Fig. 8.5
White matter plaques and cerebellar plaques in 100 μm sections. (a, b) White matter plaques usually are located close to the cortical gray matter and consist of irregularly shaped and only weakly stained flake-like deposits (a), which gradually condense into more compact forms, as seen in greater detail in (b) (68-year-old female, NFT stage III). (c, d) In phase 5, the cerebellum develops non-amyloid Aβ in the form of globules of various sizes in the granular layer (c left side) and Purkinje cell layer (d right side) or as rectangular slices (c) along dendritic trees of Purkinje cells in the molecular layer (77-year-old male, stage III). Scale bar in (d) applies to (c)
Once Aβ production begins, the total volume of insoluble Aβ plaques increases noticeably, and their number steadily increases until, apparently, a certain level is reached. Inasmuch as Aβ production continues for decades and (if at all) degradation of plaques only occurs slowly, one would expect plaques to eventually fill the entire cortical gray matter. However, in the end phase of AD, notable portions of the gray matter still are devoid of Aβ deposits even in cortical regions that are heavily laden with plaques (Fig. 8.2b). In other words, it looks as if, once a maximal plaque density has been attained, this status remains unchanged for a protracted period of time. Factors mediating the gradual reduction and final cessation of Aβ production (Hyman et al. 1993) may include the impairment and failure, over decades, of projection neurons in the non-thalamic nuclei with diffuse cortical projections. The growing presence of tombstone tangles in these nuclei would be a sign of the lost numbers of axons capable of generating Aβ.
Toxic effects on the surrounding neuropil and vessel walls are attributable primarily to Aβ when the peptide is still in a soluble and diffusible (oligomeric) form (Mucke and Selkoe 2012). However, such forms frequently evade detection in conventionally fixed tissue. Soluble forms possibly can bind to membranes not only of nerve cells but also of non-neuronal cells (Mucke and Selkoe 2012). Cellular processes from neurons and glia in the immediate vicinity of plaques can develop dystrophic processes, including accumulations of dense bodies and abnormal mitochondria. During the transition from primitive to neuritic plaques, cellular processes may even develop abnormal tau inclusions (i.e., argyrophilic dystrophic neurites). In addition, the synapses located near Aβ plaques may display signs of deterioration (Fiala 2007).
Some of the soluble and diffusible Aβ released into the ISF reaches the narrow space between the capillary wall and the end-feet of astrocytes, and it passes from there through gaps between smooth muscle cells of the vessel walls and the enveloping glia sheath. In this manner, the pathological protein drains into the regional lymph nodes of the neck similar to drainage of lymphatic fluid (Weller et al. 1998, 2009).
8.3 Transient Extracellular Aβ Deposits
At the outset of the phase when accumulations of Aβ protein that have precipitated out of the formalin fixative gradually develop out of soluble and diffusible Aβ monomers and oligomers, inconspicuous and, in part, very extensive cloud-like Aβ deposits with blurred boundaries emerge temporarily. Such deposits usually are observed in deep layers of the cortex, for instance in layer VIb of the temporal neocortex and in the deep entorhinal layer pri-γ (Fig. 8.2c, d). Faintly tinged Aβ strands develop in these layers and widely infiltrate the tissue, tending to merge into each other, and often extend into the white substance (Thal et al. 1999). There, the material gradually becomes compressed into tightly packed granules, i.e., white matter plaques (Fig. 8.5a, b) (Braak et al. 1989b).
Because of the transient nature of the cloud-like Aβ deposits, there is no readily quantifiable relationship between the depth of Aβ infiltration into the white substance and disease duration. Transient forms of Aβ deposition are congo red-negative, a fact indicating that the fibrils in the tissue are not yet cross-β sheet-rich assemblages. These transient morphological manifestations indicate that Aβ, immediately after its release into the ISF, aggregates with other Aβ molecules. The low viscosity of these formations prevents their equilibration and causes localized differences in Aβ concentrations, especially between the ISF and CSF (Englund et al. 2009).
Similarly uniform band-like Aβ formations often develop, although not inevitably, in pyramidal cell layers of CA 1, and in entorhinal layers pre-β and pre-γ (Fig. 8.2c, d). By contrast, layer pre-α generally remains free of deposits (Mufson et al. 1999). Such band-like deposits have not been reported in cases of fully developed AD and thus likely represent an intermediate type between the inconspicuous extensive cloud-like formations and the permanent spherical Aβ deposits. Only the uniform lake-like depositions of Aβ in the parvocellular layer of the presubiculum remain at this location up to the end-phase of AD (Kalus et al. 1989). The mechanisms by which the Aβ deposits develop from expansive and unsharply defined infiltrations to compact globular plaques are not known. It is possible that the migration of glial cells contribute somehow to the morphological changes undergone by Aβ deposits.
8.4 Mature Forms of Aβ Deposits and Plaque Degradation
Sharply outlined globular amyloid deposits of varying diameters represent mature forms of either primitive or cored Aβ plaques (Figs. 8.2b and 8.4b–d). They are composed of extracellular wisps of amyloid braided with assemblies of swollen dystrophic nerve cell processes (Dickson 1997b; Tolnay and Probst 1999). In contrast to primitive (diffuse) plaques (Fig. 8.4b), cored plaques possess a central amyloid mass, which is often enclosed by microglial cells and astrocytes (Fig. 8.4c). Such cores can also occur as isolated structures and in many cases are referred to as “burnt out” plaques (Fig. 8.4e, f) (Dickson 1997b; Tolnay and Probst 1999; Dickson and Vickers 2001; Fiala 2007). In contrast to transient Aβ deposits, globular plaques rarely fuse. The aggregation-prone Aβ42 is found predominantly in the core of such plaques. Dot-like initiation sites (seeds) probably lead to self-aggregation of the material and to the radial orientation of the filaments. Cored amyloid deposits can easily be mistaken for neuritic plaques (NPs) but differ from NPs by the absence of argyrophilic neuronal processes (Fig. 8.4b, d).
The various layers of the cerebral cortex display idiosyncracies locally. For example, subpial portions of layer I frequently contain confluent plaques (Fig. 8.4a), whereas layers II, IV, and Vb/VI are spared or contain only a few deposits. Spherical deposits predominate in neocortical layers III and Va. Dots are a specific feature of layer IVcα in the primary visual field (Fig. 9.8c) (Braak et al. 1989b). A few amyloid deposits also routinely occur in the white matter close to the transition to the cortex (i.e., white matter plaques) (Fig. 8.5a, b). Usually, no amyloid deposition is found in deeper portions of the white matter. Even the fiber tracts running through Aβ-laden portions of the gray matter (e.g., fornix, mamillothalamic tract, anterior commissure) only display a few precipitations. The lone exception is the perforant path that is densely decorated with Aβ plaques (Fig. 8.3) (Fiala 2007). The material in primitive plaques originally is a malleable mass that can adapt its shape to fit any structure of a given neuropil. Thus, globular Aβ deposits arise in the granular layer of the cerebellar cortex (Fig. 8.5d), while plaques in the molecular layer tend to extend vertically so as to follow the dendritic trees of Purkinje cells (Fig. 8.5c) (Braak et al. 1989b).
In the human brain, an inverse relationship exists between the degree of cortical myelination and the density of Aβ deposition. Sparsely myelinated cortical areas and layers display denser deposits than those that are rich in myelin. Densely myelinated layers, such as neocortical layers IV and Vb, which harbor the outer and inner lines of Baillarger, as well as the myelin-rich molecular layer of the allocortex remain free of Aβ deposits or show only few plaques. Areas and layers containing pyramidal cells that are rich in lipofuscin deposits also tend to show denser accumulations of globular Aβ deposits than those with sparsely pigmented neurons.
8.5 Phases in the Development of Aβ Deposits
Changes in the regional distribution pattern of plaques are less predictable than those of the intraneuronal tau inclusions, however, the gradual deposition of Aβ plaques also follows a stereotypic five-phase pattern as shown in cross-sectionally studied cases (Fig. 8.6) (Thal et al. 2002).
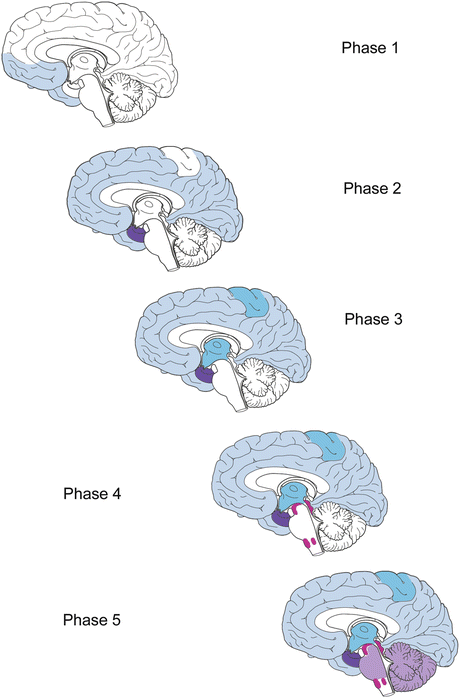
Fig. 8.6




Phases 1–5 in the development and progression of Aβ deposits. The regional distribution pattern is shown by different degrees of shading for each stage (light blue, dark blue, turquoise, magenta, purple). In phase 1, isolated plaques develop at one or more sites within the basal temporal and the orbitofrontal neocortex. Additional plaques are found in phase 2 in the allocortex and amygdala. Plaques develop in virtually all high order association areas of the neocortex. Phase 3 is marked by further expansion of Aβ plaques into secondary neocortical fields and into the striatum. They also appear in the perforant pathway and presubiculum. In phase 4, plaque formation is seen in virtually all areas of the neocortex and reaches the mesencephalon, particularly the inferior colliculi. During the final phase 5, Aβ deposition reaches the lower brainstem and cerebellar cortex
Related posts:

Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
