and Kelly Del Tredici1
(1)
Zentrum f. Biomed. Forschung AG Klinische Neuroanatomie/Abteilung Neurologie, Universität Ulm, Ulm, Germany
5.1 Stage a: The Appearance of Abnormal Tau in Axons of Coeruleus Projection Neurons
Despite the existence of a variety of neurotransmitters and modulator substances, each of the non-thalamic nuclei with diffuse ascending projections is highly susceptible to the AD-associated pathological process (Tomlinson et al. 1981; Whitehouse et al. 1981, 1985; Mann et al. 1982; Mann 1983; German et al. 1987, 1992; Zweig et al. 1988; Chan-Palay and Asan 1989; Hertz 1989; Busch et al. 1997; Rüb et al. 2000; Sassin et al. 2000; Parvizi et al. 2001; Zarow et al. 2003; Mesulam et al. 2004; Haglund et al. 2006; Grudzien et al. 2007; Weinshenker 2008; Grinberg et al. 2009; Simic et al. 2009; Braak and Del Tredici 2011; Elobeid et al. 2011; Vana et al. 2011; Trillo et al. 2013). The long and poorly myelinated axons of these nuclei might be capable of reverting to an earlier and less well differentiated state with a higher degree of tau phosphorylation more readily than heavily myelinated axons.
The AD process begins in the locus coeruleus during childhood or in young adulthood and, from there, goes on to involve other non-thalamic nuclei with diffuse cortical projections (German et al. 1992; Benarroch 2009; Sara 2009; Braak and Del Tredici 2011). The occurrence of aggregated hyperphosphorylated tau in a single coeruleus projection neuron (stage a) (Fig. 4.1b–d) may already be sufficient to pave the way for the formation of all of the succeeding lesions (stages b–VI). Many questions that arise in this context cannot be answered yet: Why does the AD-process commence in the locus coeruleus and not elsewhere? Can the process be extinguished after a spontaneous dying off or targeted removal of the initially involved neurons? Is a continual initiation over a given period of time required to fan the disease process to such an extent that it proceeds inexorably from that point on? Does the induction of the pathological process involve mechanisms and pathways that are different from those regulating its propagation?
In this context, it should be pointed out that no other neuronal system develops such abundant axonal ramifications and such close contact to the microvasculature of the CNS. Coeruleus axons regulate the microvascular system and the blood brain barrier of the CNS to the same extent that the PNS does so for the macrovascular supply to the CNS—namely, via the direct release of noradrenaline from non-junctional presynaptic varicosities (Farkas and Luiten 2001). A re-uptake of the greater part of the transmitter into the presynaptic varicosities via noradrenaline transporters occurs. Pathogens, such as viruses, which reach the vasculature or the CSF, can directly influence or penetrate into axons of the brain’s endogenous noradrenergic system (Pamphlett 2014).
Not all projection neurons within the locus coeruleus become involved simultaneously. Abnormal tau inclusions begin to develop within a few coeruleus projection neurons and then additional neurons follow suit. It is possible that this sequence is not fortuitous but predefined. We assume that the process begins in one or two nerve cells followed by additional cells on the opposite side. A delayed spread of the pathology to the non-involved side or an asymmetrical involvement pattern is relatively seldom (Fig. 2.4). The continual increase in the number of involved cells in the locus coeruleus occurs so gradually and slowly that one sees non-involved nerve cells even in end stage cases of AD. Contrary to what we would anticipate, given its very early involvement, the locus coeruleus does not become completely devoid of neurons during the lifelong AD-associated process.
Without light microscopically detectable precursors, slightly aggregated forms of hyperphosphorylated (AT8-ir) tau abruptly appear in the proximal axon of noradrenergic coeruleus neurons (Figs. 4.1b, c and 5.3b) (Braak and Del Tredici 2012). Their position is not irrelevant because functional limitations in the proximal axon will have a ripple effect well into the axon’s terminal ramifications. Obviously, the pathological material in the proximal axon is slightly less soluble in the axoplasm than monomeric tau because it does not distribute rapidly throughout the entire axon, as might be expected were the material to consist of molecules circulating freely in the axoplasm (Maeda et al. 2007; Brunden et al. 2008). Instead, the material resembles a somewhat viscous mass that moves slowly into more distally located portions (Fig. 5.3c). During formation, neither the initial axon segment nor the axon hillock nor adjoining portions of the somatodendritic compartment display any traces of abnormal material. In other words, there is no patent evidence for a transfer of abnormal tau material from the soma of involved cells into the axon. Swellings proximal or distal to the inclusions are not seen. These findings permit the assumption that the initially altered tau material in axons originates from hyperphosphorylated tau proteins of the local axoplasm.
It is frequently reported in the literature that hyperphosphorylated tau quickly relocalizes from the axon back into the somatodendritic compartment (Thies and Mandelkow 2007; Serrano-Pozo et al. 2011). Histologically, there is no evidence for such a re-sorting phenomenon, and, in fact, it seems rather improbable that, once the pathological aggregates have so suddenly appeared in the axon, sufficient amounts of diffusible hyperphosphorylated tau would be available for such a re-location.
The altered neuronal processes are straight, thin, and thread-like, and they can be found not only in the locus coeruleus (Fig. 4.1b, c) but also in the superior cerebellar peduncle (Fig. 4.1d) and at sites where the ascending catecholaminergic tract courses through the midbrain, i.e., at sites not accessible to dendrites of coeruleus melanized neurons. Moreover, the lengths of the single AT8-ir processes exceed those of the dendrites found within the locus coeruleus (Fig. 4.1d). Taken together, these findings support the interpretation that the first cellular processes to become involved are segments of proximal axons belonging to coeruleus projection neurons.
The sudden appearance of the pathological material has consequences for the cytoskeleton of the proximal axon. Abnormal changes occur in axonal microtubules, supposedly to the point of disintegration because the partially aggregated tau no longer fulfills its primary function of stabilizing the microtubules and new monomeric tau can neither be generated quickly enough in the cell body nor be transferred into the proximal axon. As a result, anterograde or retrograde axonal transport no longer occurs effectively along such badly damaged axons (Alonso et al. 2008; Higuchi et al. 2002; Mandelkow et al. 2007; Li et al. 2007; Iqbal et al. 2009). Nonetheless, the fact remains that most involved neurons survive, by means of presently unknown mechanisms, despite the presence of severe damage. Yet, such nerve cells must also be handicapped with respect to their normal functions.
At some point, the abnormal tau inclusions in the axon convert from a viscous state into a virtually insoluble polymeric mass. The short inclusions that ultimately appear in the axon are more or less of similar length and they are interspersed by AT8-immunonegative segments (Figs. 4.1e and 5.3d). This condition is stable and persists for decades: The axon can be observed to remain intact even into the final stages of the disease process despite the existence of considerable cytoskeletal abnormalities (Velasco et al. 1998). Most probably, the insoluble inclusions consist of inert and highly aggregated forms of irreversibly hyperphosphorylated tau species since they do not change in size and do not shift in either direction, nor do they show signs of re-absorption or degradation. Within AT8-immunonegative axonal segments, immunoreactions directed against axonal cytoskeletal proteins (e.g., SMI 312) are negative. At the same time, the pronounced argyrophilia of intact axons, which can be visualized in sections stained with the Campbell-Switzer silver technique (Campbell et al. 1987; Braak and Braak 1991b) is not seen in AT8-immunonegative axonal segments.
Extensive portions of abnormally altered axons can be observed in the white substance, e.g., within bundles of the catecholaminergic tract in the medulla oblongata or throughout the course of the perforant pathway. There is no evidence that these insoluble inclusions “back up” at critical junctures within the axon, such as at branching points, and the directionality of the reduced, but probably possible, transport cannot be determined from the shape of small varicosities before or behind the inclusions. Perhaps as a reactive response, acute local destruction of the axonal cytoskeleton may secondarily and transiently induce abnormally high concentrations of newly produced tau protein within the somatodendritic domain. An important light microscopic observation is that neither the viscous (but to some extent soluble) material nor their insoluble end-products in axons become Gallyas-positive. This general absence of argyrophilia fundamentally distinguishes abnormal material in axons from somatodendritic pretangle material that tends strongly to convert into Gallyas-positive fibrils.
It is unlikely that either the normal monomeric form of hyperphosphorylated tau or its aggregated and insoluble species are detrimental or toxic to the immediate surroundings. Noxious effects probably originate soley from the more viscous forms of tau (Brunden et al. 2008; Ding and Johnson 2008; Iqbal et al. 2009; Mufson et al. 2013; Tian et al. 2013) that display both a loss of physiological function (binding to microtubules) and a gain of toxic function (destruction of microtubules, impaired axonal transport). Host nerve cells are known to produce stable and insoluble species of the abnormal material to sequester the partially soluble and potentially dangerous species (Ittner et al. 2011). Thus, the abnormal and subsequently insoluble axonal inclusions remain in situ until their host neurons die while physically restricting the residual functions of axonal transport. The precise mechanisms and pathways by means of which vital transport functions in abnormally altered axons are preserved are unknown. However, residual functioning makes prolonged survival possible for many badly damaged nerve cells. The altered axon disintegrates only when the host neuron is lost, and the insoluble non-argyrophilic material then is degraded in situ without leaving behind any traces.
5.2 Stages b and c: Pretangle and Tangle Material Develops in the Somatodendritic Compartments of Coeruleus Neurons and Similar Lesions Appear in Additional Brainstem Nuclei with Diffuse Cortical Projections
The formation of abnormal material, initially similar to that observed in the axon, develops next in the somatodendritic compartment of coeruleus neurons, although the sources of this pretangle material cannot be convincingly identified at present. Owing to the development of the axonal inclusions, the axon lacks sufficient amounts of normal tau, and this situation possibly stimulates renewed tau production in the soma. However, because too few binding sites are available in the soma the newly synthesized tau probably lingers for a time in a hyperphosphorylated state in the cytoplasm. Under these conditions, it is conceivable that maximal concentrations can easily be exceeded, which then facilitate the formation of tau aggregates (pretangle material). The semantics of the term ‘pretangle material’ presume that a conversion into filamentous and argyrophilic inclusions (i.e., dendritic neuropil threads and somatic neurofibrillary tangles) is anticipated. For this reason, the term ‘non-argyrophilic abnormal’ tau is intentionally applied to the inclusions in axons that resemble pretangle material without undergoing such a conversion.
The first observable abnormal change in the somata of coeruleus projection cells consists of a transient appearance of droplet-like and probably viscous pretangle material (Figs. 5.2a–c and 5.3e) that appears to be partially soluble and, for this reason, may be highly toxic. The diameters of the droplet-like inclusions vary but clearly surpass those of somatic lipofuscin or neuromelanin granules. Large portions of the soma then become filled with this abnormal material, which surrounds both Nissl bodies and clusters of pigment granules without recognizably displacing these cellular components. The viscous pretangle material frequently spares the pale central portion of the soma. In dendritic processes, it first appears in distal branching points. From there, it fills the proximal dendritic portions and then merges with the viscous material in the soma (Fig. 5.3f).
Provided the pretangle tau persists and the involved cells do not die, the abnormal material in the somatodendritic portion tends to undergo conversion into strongly argyrophilic filamentous lesions. In addition, the material is tagged with ubiquitin, a heat shock protein required for the non-lysosomal ATP-dependent breakdown of abnormal proteins (Weaver et al. 2000; von Bergen et al. 2005; Ballatore et al. 2007; Maeda et al. 2007). Given the fact that the somatodendritic material is already aggregated at this point, so that it cannot be eliminated by the proteasome system (Kovacech et al. 2010), this ‘delayed’ ubiquitination seems futile and only ends up costing the neuron wasted energy (Garcia-Sierra et al. 2003; von Bergen et al. 2005).
Ubiquitination probably occurs shortly before the pathological material becomes argyrophilic (Lasagna-Reeves et al. 2012). The argyrophilic inclusions are selectively positive using specific silver staining techniques, such as the silver-iodide method proposed by Gallyas, which detects the lesions with great clarity (Gallyas 1971; Braak and Braak 1991b; Uchihara et al. 2001, 2005; Binder et al. 2004; Braak et al. 2006a). Neuropil threads (NTs) fill up the dendrites (Fig. 5.3h) (Ashford et al. 1998), whereas neurofibrillary tangles (NFTs) develop in the cell bodies (Fig. 5.3f, g).
There are still no answers to the question why this sequence of events is confined to the very few predisposed neuronal types described above (Table 2.1). In addition, it is unclear why involved nerve cells cannot immediately eliminate the abnormal material at a time when the molecules in question are still subject to proteasomal recycling and autophagy. Nevertheless, during this interval the viscous pretangle material remains untagged by ubiquitin (Bancher et al. 1989; Köpke et al. 1993; Uchihara et al. 2001). A dismantling or degradation of abnormal tau or signs for cellular distress, such as swelling of the neuronal soma or displacement of the Nissl material or the cell nucleus to the periphery are not observed. In other words, cell death is not imminent. On the contrary, involved nerve cells tolerate their insoluble pretangle material and, subsequently, their argyrophilic NTs/NFTs remarkably well. Thus, the viscous abnormal material is acutely toxic only initially and only briefly, whereas the insoluble material is not incompatible with long-term neuronal survival, although survival is not synonymous with optimal cell functioning.
Melanized nerve cells of the locus coeruleus that survive the first phase of pretangle material formation produce additional AT8-ir material at branching points of their dendrites. Thereafter, this material protrudes into other portions of the dendrites. Eventually, bubble-like AT8-ir varicosities develop within the dendrites (Figs. 5.2g–i and 5.3g). From these varicosities, a transition becomes evident to increasingly distorted dendrites with spherically shaped and intensely AT8-ir portions interspersed with thin thread-like segments (Figs. 5.2g and 5.3h). Distal portions of such dendrites often occur as globular structures isolated in the neuropil and separated from their stems (Fig. 5.2n). A probable consequence of these abnormalities in the dendritic trees of locus coeruleus projection neurons is the severe loss of the afferences projecting to this region, foremost among which are those of the regulatory projections from the prefrontal cortex. As a rule, such severely altered neurons die and ultimately leave behind a tombstone tangle (Fig. 9.3g, h) (Augustinack et al. 2002).
Physiologically healthy melanized locus coeruleus neurons have smoothly contoured cell bodies and only a few, relatively long dendrites (Fig. 5.3e). However, from subcortical stage b onwards, the outer somatic rim of involved cells begins to develop unusual spiked protrusions (Figs. 5.2d–f and 5.3g). These newly formed spike-like structures are slender dendritic processes with gradually diminishing diameters that are tightly packed with pretangle material (Fig. 5.2e). It is possible that melanized neurons of the locus coeruleus can eliminate some of the pathological material and/or other non-degradable inclusions (e.g., neuromelanin granules) via these spiked protrusions. Neuromelanin granules within the locus coeruleus are normally confined only to nerve cells and, for this reason, it is noteworthy that in this nucleus they are, nonetheless, also encountered extraneuronally, spaced widely apart from each other. The existence of a specialized exocytotic mechanism would explain the frequent presence of extraneuronal neuromelanin granules in young individuals.
From stage III onwards, AT8-ir changes nearby abnormal dendrites in the locus coeruleus develop in the form of a corona of thin astrocytic processes fanning out like the spokes of a wheel around the altered dendrites (Figs. 5.1c, d, 5.2o–q, and 5.3h, i). The distal portions of these astrocytic processes are enlarged and have direct contact with the pathologically altered dendritic segments. They are also intensely AT8-ir whereas the astrocytic somata are immunonegative or show only a weak reaction. As such, it appears that a degradation of the abnormal pretangle material occurs within the astrocytic processes without leaving behind any visible remnants.
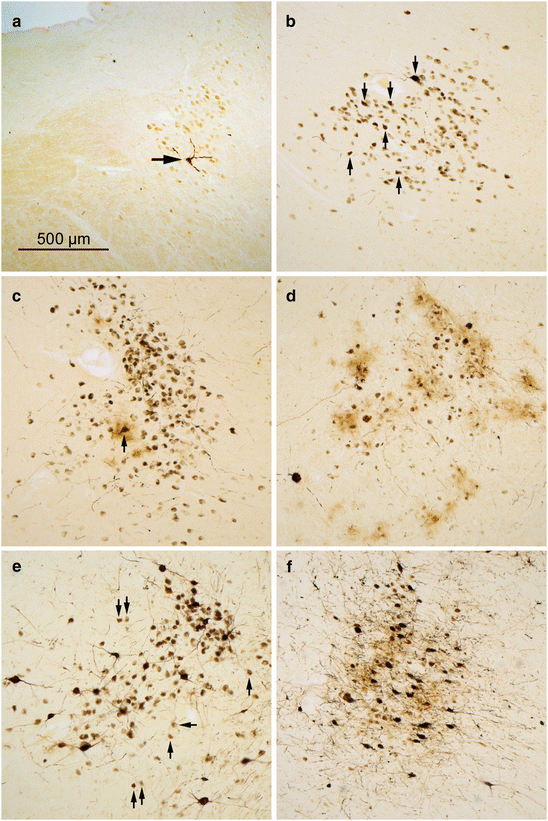
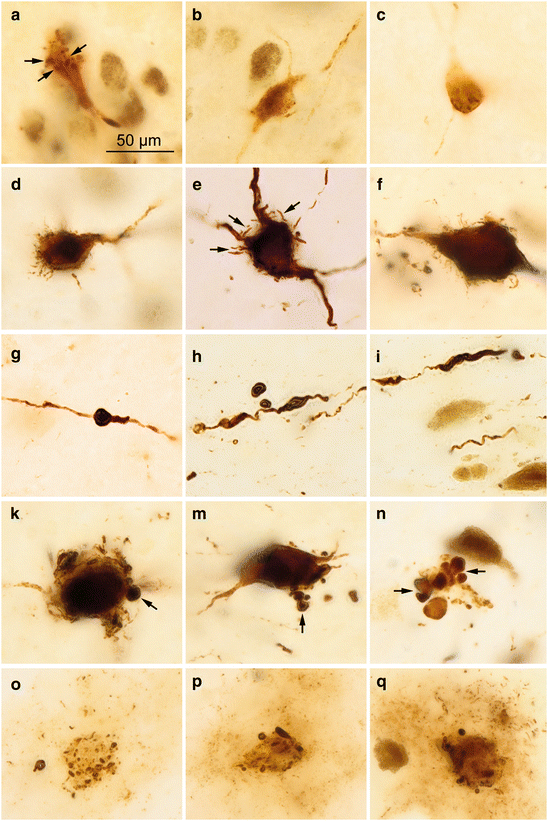
Fig. 5.1
Stages of AT8-immunopositive tau pathology in the locus coeruleus. (a) A single affected projection cell (arrow) seen in an 11-year old male (stage b). (b) Stage 1a is characterized by involvement (arrows) of multiple projection neurons (49-year-old female). (c) Affected projection cells and a single involved astrocyte (arrow) in a 68-year-old male at NFT stage I. (d) Multiple immunoreactive astrocytes (brownish ‘clouds’) and neuronal loss in 72-year-old male at NFT stage II. (e) Intact projection cells (arrows) amidst severely involved neurons accompanied by increasingly heavy neuronal loss in an 86-year-old female AD patient (NFT stage V) and (f) in an 80-year-old female with AD (NFT stage V). 100 μm sections. Scale bar in (a) also applies to micrographs in (b–f)
Fig. 5.2
 Basic Organization of Non-thalamic Nuclei with Diffuse Cortical Projections
Microtubules and the Protein Tau
Technical Addendum
The Pattern of Cortical Lesions in Preclinical Stages
Introduction
The Pattern of Lesions During the Transition to the Symptomatic Phase and in Fully Developed Alzheimer’s Disease
Basic Organization of Non-thalamic Nuclei with Diffuse Cortical Projections
Microtubules and the Protein Tau
Technical Addendum
The Pattern of Cortical Lesions in Preclinical Stages
Introduction
The Pattern of Lesions During the Transition to the Symptomatic Phase and in Fully Developed Alzheimer’s Disease




AT8-immunoreactions in locus coeruleus neurons and astrocytes. (a–c) Droplet-like inclusions (arrows) in a 21-year-old male. (d–f) Short dendrite-like AT8-ir spikes (arrows in e) develop on the formerly smooth surface of the cell soma. (g–i) Varicosities in rarified dendrites that become fragmented and occasionally detached from the main branch (h). (k–n) Towards the end of these developments, the dendrites appear as radically truncated appendages in the form of shot-like pellets (arrows). (o–q) Abnormal and weakly AT8-ir astrocytic processes surround the remnants of dead dendrites and cell somata. Micrograph in (d) is from a 20-year-old female, in (f) from an 86-year-old female. Those in (e), (g), and (n–q) are from an 85-year-old male, (h) and (i) from a 76-year-old female, (k) from a 76-year-old male, and (m), (p), (q) from a 72-year-old male. 100 μm sections. Scale bar in (a) applies to (b–q)
Related posts:
Basic Organization of Non-thalamic Nuclei with Diffuse Cortical Projections
Microtubules and the Protein Tau
Technical Addendum
The Pattern of Cortical Lesions in Preclinical Stages
Introduction
The Pattern of Lesions During the Transition to the Symptomatic Phase and in Fully Developed Alzheimer’s Disease
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
