and Kelly Del Tredici1
(1)
Zentrum f. Biomed. Forschung AG Klinische Neuroanatomie/Abteilung Neurologie, Universität Ulm, Ulm, Germany
2.1 Sporadic AD Is a Proteinopathy Linked to the Development of Intraneuronal Inclusions of Abnormal Tau Protein Which, in Later Phases, Are Accompanied by the Formation of Extracellular Plaque-Like Deposits of Amyloid-β Protein
Aggregates of abnormal proteins are the hallmarks of the AD-associated pathological process. Intraneuronal inclusions consisting of aggregated protein tau develop only in vulnerable types of CNS nerve cells (Fig. 2.1b) (Goedert and Spillantini 2006; Goedert et al. 2006; Mandelkow et al. 2007; Alonso et al. 2008; Iqbal and Grundke-Iqbal 2008; Iqbal et al. 2009; Kolarova et al. 2012; Mandelkow and Mandelkow 2012; Spillantini and Goedert 2013) whereas plaque-like deposits containing amyloid-β protein (Aβ) appear in the extracellular space of the CNS (Fig. 2.1c) (Beyreuther and Masters 1991; Selkoe 1994, 2000; Mattson 2004: Masters and Beyreuther 2006; Haass and Selkoe 2007; Alafuzoff et al. 2009; Haass et al. 2012; Masters and Selkoe 2012; Selkoe et al. 2012). Both types of proteinaceous deposits appear at different times within different CNS predilection sites and, from there, spread out systematically into previously uninvolved brain regions. In the course of the pathological process, both types of lesions increase in severity. The AD process begins with intraneuronal aggregations of the protein tau followed, after a time-lag of approximately a decade, by the extracellular deposition of Aβ (Braak and Braak 1997b; Duyckaerts and Hauw 1997; Silverman et al. 1997; Braak et al. 2013) (Fig. 9.16, Tables 9.2 and 9.6). Neuritic plaques (NPs), i.e., combined deposits consisting of insoluble Aβ with dystrophic neurites that contain aggregated tau, develop only in the late phase of the disease process (Braak and Braak 1997b; Braak et al. 2013).
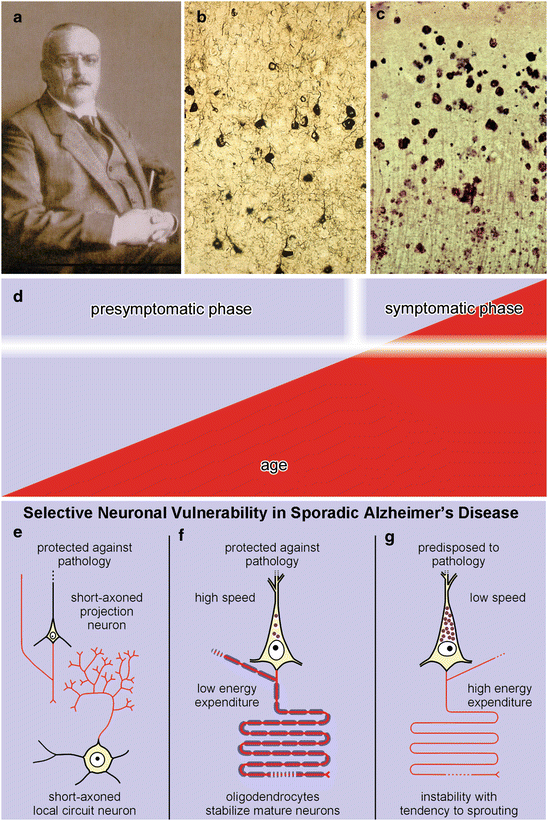
Fig. 2.1
(a) Alois Alzheimer. (b) Neurofibrillary lesions shown by Gallyas silver-iodide staining technique. (c) Aβ plaques visualized by Campbell-Switzer silver-pyridine staining technique. (d) The lesions (red) increase in severity and extent without remission during a long presymptomatic disease phase that reaches a shorter and final symptomatic phase. (e–g) Selective neuronal vulnerability in sporadic AD. Neuronal types protected against AD-associated pathology include short-axoned projection cells and local circuit neurons (e) and pyramidal cell with a long and heavily myelinated axon (f). Vulnerable types of pyramidal cells have a long and sparsely myelinated axon (g). (e–g adapted with permission from H Braak and K Del Tredici, Adv Anat Embryol Cell Biol 2009;201:1–119)
In contradistinction to the pathology that emerges in the course of sporadic Parkinson’s disease (PD) (Braak and Del Tredici 2009; Del Tredici et al. 2010; Del Tredici and Braak 2012), the hallmark lesions associated with AD remain almost exclusively confined to the CNS. Nerve cells of the peripheral and enteric nervous systems (PNS, ENS) likewise contain normal tau and the amyloid precursor protein (APP), but only the olfactory epithelium is known to develop abnormal protein aggregates (Arnold et al. 2010; Kovács 2013). Why is it that the ENS and other PNS sites do not develop aggregated tau and Aβ? Attempts to understand the pathogenesis of the hallmark lesions should also explain why the AD process develops almost exclusively within the CNS.
The AD-associated pathological process, once started, is not known to regress, improve spontaneously, or go into remission. This controversial but important aspect of the AD process is treated below in Sect. 2.3. The process spans nearly a lifetime unless it is interrupted by death from other causes. In other words, it encompasses a much longer time-span than the clinically recognizable phase and consists of a relatively short symptomatic final phase and a long preclinical phase. The last phase, accompanied by the loss of cognitive and executive functions, manifests itself, as a rule, only at an advanced age (Fig. 2.1d).
The hallmark lesions are generally accompanied in this late phase by pathologies attributable to disorders that develop with increasing age (e.g., vascular disease, metabolic syndrome, concomitant neurodegenerative diseases) that aggravate, to varying degrees, the clinical picture, making the diagnosis and the questions surrounding the pathogenesis of AD increasingly complex (van Gool and Eikelenboom 2000; Iadecola 2004, 2010; Fotuhi et al. 2009; Duyckaerts et al. 2009; Grinberg and Thal 2010; Chui et al. 2012; Hunter et al. 2012; Korczyn et al. 2012; Korczyn 2013; Thal et al. 2012; Wang et al. 2012; Kovács et al. 2013; Serrano-Pozo et al. 2013). The co-occurrence of sporadic AD and the lesions associated with sporadic PD or frontotemporal lobe degeneration (FTLD) is especially problematic (Duyckaerts et al. 2009; Mesulam et al. 2014). Nevertheless, owing to the co-occurrence of multiple pathologies, each case is unique. For this reason, sporadic AD is not always viewed as a single disease entity but as a syndrome leading to dementia (Korczyn 2013).
In youth and in early adulthood Aβ plaque deposition is non-existent or rare. Tau aggregates, on the other hand, occur before puberty and are absent only in very young children (Fig. 9.12) (Braak et al. 2011; Braak and Del Tredici 2011, 2012). The ongoing development of both the intraneuronal tau aggregates and the extracellular plaque-like Aβ deposits is extraordinarily slow, so that the hallmark lesions cannot be said to originate only in old age or typically during aging (Morrison and Hof 1997; Nelson et al. 2011). Nonetheless, clinically overt AD has been viewed as a disorder that is intrinsic to the aging process or, at the very least, indirectly attributable to it. Age-related factors capable of damaging aging postmitotic cells, such as chronic inflammation, oxidative stress, mitochondrial and metabolic dysfunction, blood-brain barrier impairment, or failure of the ubiquitin-proteasomal system, are viewed as pivotal factors in the pathogenesis of AD (de la Monte and Tong 2013; Pohanka 2013; Yan et al. 2013; Arshavsky 2014). None of these factors alone, however, suffices to explain why the hallmark lesions consistently and selectively develop in only a few types of nerve cells. Obviously, they fail to affect all known types of postmitotic cells inside and outside of the CNS. Even when the discussion is confined to the CNS, it is evident that AD does not involve all neuronal types there indiscriminately. Rather, the AD-process is a remarkably selective one in that it develops in only a minority of neuronal types while sparing all of the rest. In addition, the effects of age-related factors do not explain why the tau lesions develop in children and young adults during the early phase of the disease. Thus, advanced age per se is not necessary for the formation of the AD-related tau lesions: The pathological process underlying sporadic AD is not ‘age-dependent’ but an uncommonly protracted and progressive process that frequently extends into old age (Fig. 2.1d). Clinical symptoms develop subtly, and nerve cell impairment leads to a gradual loss of fundamental functions that first appear after a given threshold is exceeded (Fig. 2.1d).
The present monograph rests upon the assumption that it is the pathological process depicted above which, in its final phase, causes the clinical symptoms of AD. Individuals with a history of cognitive dysfunction, whose brains do not have the hallmark AD lesions, should be classified in the heterogeneous group of non-AD dementias (Tolnay and Probst 1999; Clavaguera et al. 2013a; Dickson et al. 2007).
2.2 Some Neuronal Types Exhibit a Particular Inclination to the Pathological Process While Others Show a Considerable Resistance To It
The deterioration of the CNS in sporadic AD specifically targets predilection sites in select subcortical nuclei and cortical areas. Of the numerous neuronal types within the CNS, only some develop AD-related pathology, whereas others, including those directly in the vicinity of involved nerve cells, remain morphologically and functionally intact (Braak and Braak 1999). The resultant non-random regional distribution pattern of the lesions is reflected by dysfunction of select neuronal types and, in some regions, mild neuronal loss. The pathological process selectively affects high-order processing regions (Arendt 2000). However, because these regions are not absolutely essential for survival the disease process can exist and progress for nearly an entire lifetime until death, which is usually caused by severe autonomic dysregulation.
Most of the vulnerable neuronal types are phylogenetically late-appearing elements that also achieve functional maturity late in life (Rapoport 1988, 1989; Bartzokis 2004; Rapoport and Nelson 2011). These nerve cells frequently retain a high degree of structural plasticity in the adult brain and show signs of immaturity that endure well into adulthood (Stephan 1983; Rapoport 1990, 1999; Arendt et al. 1998; Arendt 2000; Bufill et al. 2013). The distalmost segments of their dendrites often display a slow and ongoing growth pattern that persists even when dendritic maturation in non-susceptible nerve cell types is already complete (Arendt 2000). In addition, the vulnerable cells frequently display a loss of regulation over neuronal differentiation with a partial reactivation of the cell cycle (Arendt 2004, 2005, 2012).
Nearly all of the diverse types of nerve cells in the CNS that are prone to develop AD-related tau aggregates are projection neurons, i.e., nerve cells with a localized dendritic arbor and an axon that is disproportionately long in relation to the size of the host cell soma (Fig. 2.1f, g). Inasmuch as glutamatergic, gabaergic, dopaminergic, noradrenergic, serotonergic, histaminergic, and cholinergic projection cells become involved, the type of neurotransmitter or neuromodulator synthesized is not essential for predicting which nerve cells are especially vulnerable or predisposed to the AD process. Short-axoned neurons generally do not become affected (Fig. 2.1e) (Braak and Braak 1999; Benarroch 2013) with the rare exception of large cholinergic local circuit neurons in the striatum and chandelier cells in the basal temporal neocortex (see Sect. 9.2). In addition, nearly all projection neurons with a short axon remain intact, such as the small pyramidal cells in neocortical layers II and IV (spiny stellate cells) (Fig. 2.1e) (DeFelipe et al. 2002) and those of the presubicular parvocellular layer. An aggrecan-based perineuronal net surrounds subsets of cortical and subcortical projection neurons, and these subsets do not develop intraneuronal tau lesions. As a result, perineuronal nets may contribute to the selective resistance of these nerve cells against the tau-mediated pathological process (Morawski et al. 2010). The overall resistance on the part of short-axoned neurons has its consequences: When a projection essentially synapses only on local circuit neurons—e.g., as is the case with neocortical pyramidal projections from layer V to layer III and those from layer VI to layer IV (Bannister 2005)—the pathological process cannot spread any further. The potential route of propagation from projections of the anterior subnuclei of thalamus to the presubicular parvocellular layer also comes to a halt at the short-axoned neurons there.
All of the endangered neuronal types generate a long and thin-caliber axon that either is encased by a thin myelin sheath or does not undergo myelination (Fig. 2.1g). Projection neurons reach full functional maturity only after their axons have achieved their stipulated degree of myelination (van der Knaap et al. 1991). Human neocortical projection neurons in prefrontal or in high-order sensory association areas commence myelination late in life and, thus, are thinly myelinated and predisposed to the AD-related process (Bartzokis 2004). By contrast, cortical or subcortical projection neurons with heavily myelinated axons resist developing tau aggregates (Fig. 2.1f). These include the Betz cells in the primary motor area, Meynert’s pyramidal cells in the striate area, or host neurons of the medial longitudinal fascicle.
Increased thickness of the myelin sheath provides greater velocity of axonal conduction and, at the same time, a considerable reduction of the metabolic demands placed on the host cell for the transmission of the impulses (Fig. 2.1f). By contrast, rapid-firing projection neurons with an unmyelinated or immaturely myelinated axon are subject to higher energy turnovers and are thereby chronically exposed to oxidative stress (Fig. 2.1g) (Pohanka 2013; Yan et al. 2013). The relatively postponed onset of myelogenesis in neurons of high-order regions which are not absolutely essential for preservation of basic brain functions results in high energy consumption (Fig. 2.1g). Yet, it is precisely these neurons that enrich and optimize complex activities, such as learning, memory, and perception, that are particularly prone to develop AD lesions (Arendt 2000). These include late-maturing pyramidal cells, whose axons preferentially develop connections to the distalmost dendritic segments of other cortical pyramidal cells but have no immediately obvious functions.
The myelin sheath provides a mechanical barrier against viruses and other pathogens by virtually isolating the axon from the surrounding extraneuronal space. However, this also means that the energy supply for long axons cannot originate in the cell soma. Instead, glial cells increasingly assume this function. The axon is embedded among oligodendroglial cells that protect and support it. Local astrocytes take up substances critical for the energy balance via contacts to the cerebral vasculature and redistribute them to oligodendrocytes by means of gap junctions. Additional mechanisms enable the transfer of these substances from the oligodendrocyte to the axon (Nave 2010; Lee et al. 2012b).
Nerve cells of the human adult generally are richly supplied with lipofuscin or neuromelanin granules (Braak 1980; Double et al. 2008) and, notably, all of the vulnerable cell types contain such granules (Fig. 2.1g). The presence of lipofuscin or neuromelanin deposits alone, however, does not suffice to account for the susceptibility of projection cells to the AD-process because many of them develop large amounts of these paraplasmatic granules with advancing age without developing tau aggregates, as, for instance, the Betz cells of the motor cortex or projection neurons of the lateral geniculate body. On the other hand, nerve cells that conspicuously lack lipofuscin or neuromelanin granules, or that contain only a few granules even in old age, consistently resist the pathological process. Prime examples are the large projection neurons of the hypothalamic lateral mamillary nucleus where, even in old age, lipofuscin granules are hardly present and AD-associated tau aggregates do not develop.
Viewed against this background, the combination of paraplasmatic pigment granules and an unmyelinated or sparsely myelinated long and thin-caliber axon in phylo- and ontogenetically late-maturing projection neurons appears to be a deficiency of the human CNS that may be necessary for the induction of the AD-associated process (Fig. 2.1g) (Braak and Braak 1999).
2.3 Consistent Changes in the Regional Distribution Pattern of Intraneuronal Inclusions Make a Staging Procedure Possible
As in other illnesses, at some point during the pathological process, patients cross a threshold from the preclinical phase to the symptomatic manifestation of AD (Fig. 2.1d). By the time clinicians make their diagnosis, patients are, relatively speaking, in the late phase of a larger pathological process. The disease festers in the CNS for decades until its dimensions are such that dysfunctional behavior becomes manifest (Dubois et al. 2010).
Cases with clinically recognizable symptoms usually can be assigned to one of four neuropathological subgroups (neurofibrillary stages III, IV, V, VI), which differ with respect to the topographic extent of the AD-related tau pathology (Fig. 2.2a, e). The idea of regional expansion rests on the assumption that the lesions most likely develop in a consecutive manner within the CNS and then increase in severity and extent (Fig. 2.2b). Each subgroup, therefore, displays newly affected regions in addition to the tau lesions existing at previously involved sites (Braak et al. 2011).
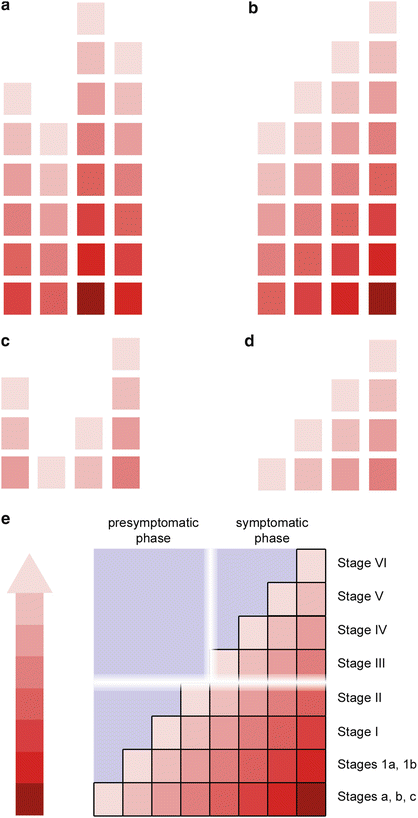
Fig. 2.2
Presymptomatic and symptomatic phases of sporadic AD. (a) Most symptomatic cases with AD-associated tau pathology fall into four subgroups. (b) Given the consistency of this finding and, based on the topographic distribution pattern of the lesions, the four groups can be arranged to show disease progression (neurofibrillary stages III, IV, V, VI). (c) Similarly, most non-symptomatic cases also fall into four subgroups (d) and can be ordered sequentially (pretangle stages a–c, 1a/1b, and NFT stages I, II). (e) Mild to moderate tau lesions develop over time until a threshold from the prodromal to the symptomatic (clinical) phase is crossed. Roman numerals correspond to stages of Gallyas-positive (argyrophilic) lesions. Arabic numerals represent stages with cortical AT8-ir (non-argyrophilic) lesions (stages 1a/1b), whereas lower case letters designate stages with subcortical AT8-ir non-argyrophilic lesions (stages a–c). See also Table 2.1
Tau aggregates occur as incidental findings in non-symptomatic individuals (Linn et al. 1995; Dubois et al. 2010; Ferrer 2012), and these also can be divided into one of four subgroups (Fig. 2.2c, d). These lesions sometimes are viewed as a variant of neuronal aging, neuroprotective, or as possible markers of a non-AD-related tauopathy (Attems et al. 2012; Cower and Mudher 2013; Jack and Holtzman 2013; Jack et al. 2013; Thal et al. 2013; Braak and Del Tredici 2014; Crary et al. 2014; Kuchibhotla et al. 2014). However, they also can be interpreted as markers of early phase disease—comparable to malignant cells in a carcinoma that fail to produce symptoms but mark the onset of a pathological process (Fig. 2.2e). The concept that incidental tau aggregates are completely ‘normal’ requires the highly problematic definition of the point at which such tau inclusions convert from a ‘normal’ status into ‘disease-related’ lesions. Disease-related lesions existing prior to the clinical manifestation of a disease are usually regarded as prodromes. As a clinical entity, AD includes dementia, but the AD-related pathological process includes a very protracted preclinical phase, which certainly occupies a pivotal position in relation to the pathogenesis of AD (Figs. 2.1d and 2.2e).
We view such clinically mute incidental tau lesions as a potential threat to the CNS for the following reasons: The presymptomatic and symptomatic disease phases are both marked by the presence of the same types of intraneuronal tau aggregates in the same types of nerve cells and at the same regional predilection sites. Second, since the lesional pattern of the last preclinical subgroup closely resembles that of the first symptomatic subgroup (compare Fig. 2.2d and b), both sets of subgroups combined can be taken to reflect the full spectrum of the pathological AD-process (Fig. 2.3a–c). The lesions develop in a remarkably predictable and consistent sequence across cases (Figs. 2.2e, 9.8, and 9.13) (Kemper 1984; Arnold et al. 1991; Braak and Braak 1991a, 1997a, b, 1999; Braak et al. 2006a; Hyman and Gómez-Isla 1994; Duyckaerts and Hauw 1997; Delacourte et al. 1999). Third, the existence of asymmetric tau distribution patterns, as seen in double hemispheres sections immunostained for AT8, indicates that a neurobiological continuum of tau pathology exists and that this pathology also tends to progress within one and the same individual, albeit at a different pace (Fig. 2.4a–c).
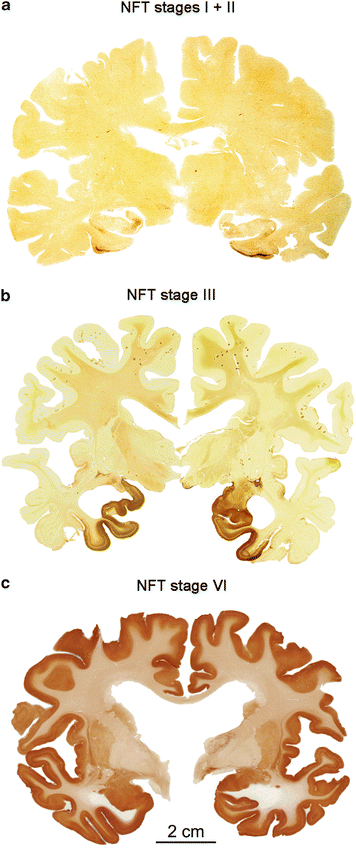
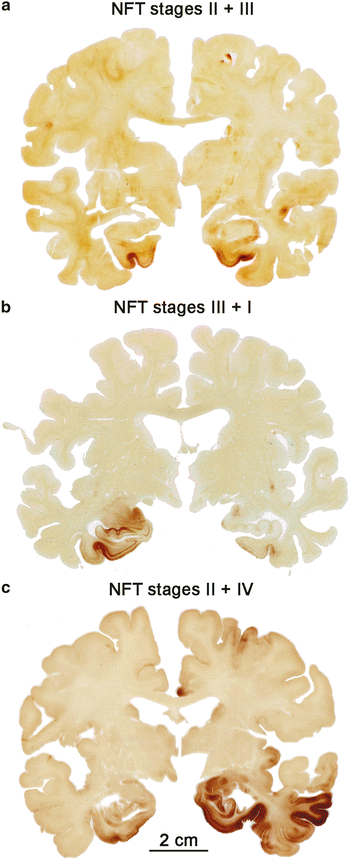
Fig. 2.3
Overview of AD-related AT8-immunopositive tau stages in three double hemispheres of 100 μm thickness. (a) The hemispheres of this cognitively intact 80-year-old female display NFT stages I (left) and II (right) in the absence of Aβ plaques. (b) NFT stage III in a 90-year-old female, who died of a malignant pancreatic neoplasm. Aβ plaques were also present. (c) Frontal section from a severely demented 72-year-old female AD patient (cause of death aspiration pneumonia) with bilateral NFT pathology and ventricular widening typical of stage VI. Aβ plaques were also present (adapted, in part, with permission from H Braak and K Del Tredici, Alzheimers Dement. 2012; 8:227–233). Scale bar in (c) is valid for (a) and (b)
Fig. 2.4
Double hemispheres of 100 μm thickness showing asymmetrical AD-related AT8-immunopositive tau stages. (a) Section from a cognitively intact 77-year-old female (cause of death myocardial infarction) at NFT stage III (left) and stage II (right). No ventricular widening is detectable. (b) Frontal section from a 75-year-old female patient (cause of death chronic lymphatic leukemia) with NFT stages III (left) and NFT stage I (right). (c) This frontal section from a cognitively impaired 75-year-old male (cause of death metastatic pulmonary neoplasm) displays pathology corresponding to NFT stages II (left) and IV (right). Deviations of more than one NFT stage are unusual (compare Fig. 2.3). See also Sect. 9.3. Scale bar in (c) applies also to (a) and (b)
Locating the first tau aggregates in an organ as voluminous as the human CNS might seem like an insurmountable undertaking, but it is possible provided the predilection sites of the pathological process are known. The lesions do not develop randomly, here at one site, there at another. Instead, the AD process is stereotypic, beginning in the same regions and advancing with little inter-individual variation. As a rule, one does not see appreciable differences bilaterally with respect to the topographic distribution of tau pathology in double hemisphere sections (Fig. 2.3), and when discrepancies occur, they do not amount to more than one (Figs. 2.3a and 2.4a) or two stages (Fig. 2.4b, c). In cases with advanced stages, this phenomenon disappears and the hemispheres of such cases display a more or less symmetric involvement (Fig. 2.3c). In this context, it should be noted that the concept of neuropathological staging is recommended for practical reasons only. In principal, it is an artificial construct because, as pointed out above, the hallmark lesions develop continually rather than in definite steps (Braak and Braak 1991a).
Related posts:
 Basic Organization of Non-thalamic Nuclei with Diffuse Cortical Projections
Microtubules and the Protein Tau
Early Presymptomatic Stages
Technical Addendum
The Pattern of Cortical Lesions in Preclinical Stages
The Pattern of Lesions During the Transition to the Symptomatic Phase and in Fully Developed Alzheimer’s Disease
Basic Organization of Non-thalamic Nuclei with Diffuse Cortical Projections
Microtubules and the Protein Tau
Early Presymptomatic Stages
Technical Addendum
The Pattern of Cortical Lesions in Preclinical Stages
The Pattern of Lesions During the Transition to the Symptomatic Phase and in Fully Developed Alzheimer’s Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


