Diagrams iIlustrating the gradual loss of white matter with aging. The left panel shows the trajectory of MRI frontal lobe white matter volume, and the right panel depicts the myelin staining of intracortical white matter (from Bartzokis 2011).
A key feature of this hypothesis is that amyloid may be toxic only late in the course of AD pathogenesis, after the early phase when it may in fact represent a protective response to insults such as ischemia, TBI, oxidative stress, and inflammation (Castellani et al., 2008; Catellani and Perry, 2014). Experimental work, for example, has shown that APP knockout mice have increased mortality in response to ischemia, suggesting that APP and its cleavage fragments have a protective role in response to this form of stress (Koike et al., 2012). Thus, for a time, the many insults of aging can be compensated for by the APP, Aß, and possibly other repair mechanisms, and only after a critical point is this capacity eroded to the point where cortical amyloid accumulates to a level at which it is neurotoxic. Tau pathology also develops (Querfurth and LaFerla, 2010), and by this time the progressive dementia of AD typically becomes apparent. Amyloid plaques and tau tangles are abnormal, but rather than be considered primary causes of dementia, they are better considered the late sequelae of a long struggle of the normal brain, over perhaps many decades, to stave off the many pathologies of aging that primarily beset the white matter.
Thus, in the myelin model, cortical involvement is important in AD, but not primary, and the deposition of abnormal proteins is downstream of the fundamental white matter neuropathology that has occurred many years before. When amyloid and tau accumulate to the point at which they do become toxic, protein deposition is accompanied by massive synapse loss and neuronal dropout in relevant cortical regions, and these phenomena are thought to be the basis for the clinical deficits of the disease (Terry et al., 1991; DeKosky, Scheff, and Styren, 1996; Coleman and Yao, 2003; Koffie, Hyman, and Spires-Jones, 2011).
Recent information has suggested that synapse loss may primarily result from the toxic effect of Aß oligomers, which are recognized to colocalize with neuritic plaques, and neuronal loss may follow through the toxic effects of hyperphosphorylated tau (Koffie, Hyman, and Spires-Jones, 2011). The subsequent clinical progression of AD to its terminal state may then be a result of unchecked protein propagation throughout the cerebral cortex. In this regard, Prusiner (2012) has raised the provocative idea that amyloid propagates much like a prion. This notion is compatible with the myelin model in that early insults to myelin may instigate the neuropathological cascade that eventually destroys the cerebral cortex via the progressive spread of amyloid and tau toxicity.
The prominence of white matter injury in the early pathogenesis of AD has been substantially supported by neuroimaging. This work has mainly been done with older persons who harbor ischemic white matter lesions. The deleterious cognitive impact of white matter hyperintensity progression has been often confirmed (Gunning-Dixon and Raz, 2000; Kloppenborg et al., 2014), and it appears that progression of periventricular white matter lesions can induce cortical atrophy (Kloppenborg et al., 2012). Compared with normal older people, those with both AD and mild cognitive impairment (MCI) have a greater number of white matter hyperintensities, reduced cerebral white matter volume, and widespread microstructural white matter disease as determined by diffusion tensor imaging (DTI) (Radanovic et al., 2013). Studies of older people have shown that white matter hyperintensity volume predicts the appearance of both MCI (Silbert et al., 2012) and AD (Brickman et al., 2012b), whereas hippocampal volume loss predicts neither. Studies such as these offer an emerging perspective that early white matter abnormalities related to common neuropathological processes may in some way drive the later cortical neuropathology of AD.
In view of considerable evidence that AD cortical neuropathology begins in the medial temporal regions, including the hippocampus and entorhinal cortex (Querfurth and LaFerla, 2010), direct examination of white matter in these areas has been particularly informative. The fornix has attracted much recent attention because of its critical position as the major efferent tract of the hippocampus (Oishi and Lyketsos, 2014; Nowrangi and Rosenberg, 2015), and this structure has special appeal as a potential early site of AD pathogenesis in view of the dominance of recent memory dysfunction as the usual presenting feature of the disease. Longitudinal study combining MRI and DTI in normal elders who converted to AD has indeed disclosed that the fornix may be vulnerable to deterioration before medial temporal gray matter is affected (Fletcher et al., 2014). The myelin model predicts that early white matter damage precedes cortical disease, and indeed the fornix is susceptible to many common forms of white matter pathology, including ischemia and TBI (Thomas, Koumellis, and Dineen, 2011). Typical small vessel disease of older adults as seen by MRI can damage the fornix (Moudgil et al., 2000), and DTI studies have documented delayed loss of forniceal white matter integrity after TBI (Adnan et al., 2013). Forniceal DTI changes have even been observed in presymptomatic carriers of familial AD mutations (Ringman et al., 2007). These studies add credence to the myelin model by documenting the neuropathological vulnerability of a key limbic tract that could explain the predilection of early AD for the medial temporal region.
The myelin model also sheds light on another long-recognized clinical feature of AD, which is that the disease is more common in women, even after accounting for the greater longevity of women compared to men in industrialized societies (Thies et al., 2013). Postmenopausal estrogen deficiency has been postulated as a risk factor of AD, although clinical trials with estrogen in women with AD have proven unsuccessful. Recent MRI studies of normal elders have shown that white matter hyperintensities are more common in women, which may mean that estrogen deficiency contributes to brain ischemia and the development of white matter lesions (de Leeuw et al., 2001; Sachdev et al., 2009). Thus the higher prevalence of AD in women could be mediated by an increased risk for white matter hyperintensity development after menopause, which in turn leads to the etiopathogenetic sequence predicted by the myelin model.
Further perspective on this idea can be gleaned from considering the notion of MCI (Petersen et al., 1999) in more detail. This influential and much-studied condition has been widely regarded as a transitional stage between normal aging and AD, as many people with MCI do progress into dementia after variable intervals. If MCI is indeed the precursor of AD, and the amyloid hypothesis is correct, it would follow that plaques and tangles would be present in MCI but less abundantly so than in AD. Yet this is not always the case. Whereas many people with MCI do have AD changes, others do not, and indeed a host of other factors such as vascular pathology, Lewy body deposition, inflammation, oxidative stress, mitochondrial dysfunction, changes in genomic activity, and disrupted metabolic homeostasis can be found (Stephan et al., 2012). It thus appears that MCI is a heterogeneous condition that does little to support the amyloid hypothesis. Rather, MCI may more accurately be regarded as a pre-dementia state reflecting the brain in the midst of many diverse processes, with the clinical state representing the balance between the detrimental effects of these factors and the brain’s capacity to withstand these insults.
In the myelin model, amyloid could be seen as one of the protective mechanisms employed by the brain – one that helps repair myelin and, in the course of doing so, leaves behind cortical amyloid as evidence that this process has occurred. Recent neuropsychological investigations of asymptomatic persons with AD neuropathology have been revealing in this context. In a study of asymptomatic people who died within 2 years of neuropsychological assessment, Monsell and colleagues (2014) found that those who were found to have AD changes displayed greater attentional and working memory deficits than those who had no AD changes. Whereas this finding could be interpreted as documenting the early effects of AD neuropathology in the cortex, it could alternatively be seen as evidence that the brain is responding to age-related damage to cerebral myelin. The latter interpretation would hold that attentional and working memory deficits in fact reflect white matter pathology, and the cortical amyloid is evidence of myelin repair.
When the dementia of AD ultimately sets in, the case can be made that the cortical synapse and neuronal loss reflect the damaging impact of progressive amyloid and tau deposition. From the perspective of the myelin model, it would follow that the clinical features of AD would result from the inability of the brain to continue myelin repair, and hence there develops an overwhelming accumulation of cortical amyloid and then tau. As this process is protracted, however, and may involve many years, it can be imagined that fluctuations in the neurotoxicity of these proteins occur as the disease progresses. An interesting idea has been suggested that the neuropathological alterations in the cortex may correspond to well-recognized clinical fluctuations of AD patients, reflecting a dynamic process of chronic protein intoxication that the brain can temporarily overcome (Palop, Chin, and Mucke, 2006). This notion may imply that even when cortical neuropathology is clinically eloquent, some opportunity for intervention may yet be possible (Palop, Chin, and Mucke, 2006).
The myelin model of AD is most pertinent to sporadic AD, which typically occurs in late life and implicates a wide range of environmental factors (Box 13.1). Yet the genetics of AD deserves attention in light of available data on the AD susceptibility gene APOE, early-onset familial AD, and DS. Remarkably, early white matter damage consistent with the myelin model can be implicated in all of these conditions. First, in contrast to the widely held assumption that the APOE ε4 genotype increases AD risk by enhancing Aß production, this allele may alternatively hinder myelin repair because of impaired cholesterol transport (Bartzokis, 2011). As discussed in the previous chapter, neuroimaging studies have shown that cognitively normal APOE ε4 carriers have lower MRI white matter volume that correlates with slower processing speed (Ready et al., 2011), decreased frontal white matter fractional anisotropy (FA) on DTI that correlates with lower executive function (Ryan et al., 2011), and disrupted structural connectivity together with diminished memory performance (Chen et al., 2015). In contrast, APOE ε2 carriers, who are presumably protected from the effects of white matter damage because of more efficient delivery of cholesterol and lipids to axons (Bartzokis, 2011), have higher FA in many white matter tracts (Chiang et al., 2012).
Next, mutations associated with familial AD may produce disease by virtue of inadequate myelination or myelin repair, and the deposition of cortical Aß may not be a crucial early event (Bartzokis, 2011). Supportive evidence for this notion comes from the DTI work of Ringman and colleagues (2007) documenting white matter changes in presymptomatic presenilin-1 and APP mutation carriers.
Finally, with regard to DS, it is of considerable interest that, despite developing AD pathology, some DS patients do not become demented (Tyrrell et al., 2001). Given this observation, one can imagine that Aß does not necessarily exert a pathogenic effect, and that damage to the brain white matter may be more critical. It has long been known that white matter hyperintensities are common in adults with DS, even before dementia occurs (Emerson et al., 1995), and may reflect vascular or other white matter insult. More recently, a provocative DTI study of adult DS patients with and without dementia found that frontal white matter integrity was diminished in those with dementia (Powell et al., 2014). These data merit particular attention in view of the observation that dementia in DS appears to be characterized by personality and behavior changes along with executive dysfunction, all securely associated with frontal lobe involvement (Ball et al., 2006). Thus it may be that the primary insult in DS leading to dementia is actually damage to white matter, which may then activate Aß deposition that reflects an attempt at brain repair after white matter injury.
When considered in its entirety, perhaps the most interesting implication of the model is a more refined view of amyloid in the complex etiopathogenesis of AD. Whereas plaques and tangles may be central to the pathology of AD, it is not clear that they hold the same position with respect to pathogenesis (Castellani and Perry, 2014). Rather than being seen as uniformly toxic, amyloid may be more usefully conceptualized as part of an adaptive or protective system to deal with encroaching changes of brain aging and the insults accumulated over a lifetime. In this context, it is appropriate to point out that while clinicians and neuroscientists are quite logically drawn to readily observable histologic hallmarks of a disease such as AD – much as they are to the clearly undesirable pathologies such as thrombosis, infection, and neoplasia – it is not at all clear that plaques and tangles have the same destructive impact on the brain as do the better-known pathologies with which medicine is well acquainted. The normal functions of APP and Aß remain unknown, and the assumption that the mere presence of neuritic plaques guarantees their pathogenicity in all cases is premature. Rather, it may be more accurate to consider a more diverse function of these proteins, even to a point where APP and Aß may be beneficial at some point in disease development and toxic only later.
Implications for prevention and treatment
The myelin model of AD offers a strikingly novel idea about how this devastating disease begins. If the model is correct, many new and important implications for prevention and treatment should be recognized. Two principal topics merit consideration in this light: (1) the prediction that the major clinical trials now underway for the treatment of AD are not likely to succeed, and may even be hazardous, and (2) the prospects for alternative treatment modalities that may hold more promise.
The continuing influence of the amyloid hypothesis has led to large-scale clinical trials in AD designed to rid the brain of amyloid. At present, this approach relies heavily on passive immunization with monoclonal antibodies that can clear presumably toxic Aß from the brain (Prins and Scheltens, 2013). Although amyloid-targeted therapies have uniformly failed to date, trials using such monoclonal antibodies as solaneuzumab are being conducted in AD, and even in elders with “preclinical AD” who have brain amyloid demonstrated by florbetapir PET but are cognitively normal (Prins and Scheltens, 2013). While the idea of removing Aß before dementia sets in has some plausibility, it is not clear that Aß is toxic in nondemented individuals, as it is well known that at least 25% of normal older people have sufficient brain amyloid at autopsy to qualify as having AD (Gandy and DeKosky, 2013).
Moreover, removing the protein at an early stage may actually be harmful because of interference with normal repair processes that are underway before dementia begins (Bartzokis, 2011; Castellani and Smith, 2011). Other concerns relate to the propensity of these agents to produce adverse neuroimaging findings, known as amyloid-related imaging abnormalities (ARIAs), that include vasogenic edema, microhemorrhages, hemosiderin deposition, and superficial siderosis (Prins and Scheltens, 2013). The etiology of these ARIAs is thought to be increased vascular permeability following clearance of amyloid from brain arterioles (Prins and Scheltens, 2013), and the use of agents with these risks is concerning, particularly in individuals who are clinically normal.
Another therapeutic approach to AD based on the amyloid hypothesis is the exploitation of ß-secretase inhibition (Vassar, 2014). ß-secretase, or, more specifically, the ß-site APP cleaving enzyme (BACE), is the enzyme initially involved in the generation of Aß from its parent molecule APP, and inhibition of this enzyme has been proposed as an AD treatment that could succeed by preventing the formation of Aß (Vassar, 2014). From the perspective of the myelin model, the important point regarding ß-secretase is that this enzyme also activates neuregulin, a protein growth factor important in the process of myelination (Xu et al., 2006). This topic was considered in Chapter 13 with respect to myelin repair, where it was pointed out that the promyelinative activity of neuregulin has been observed in the brain as well as in the peripheral nervous system (Hu et al., 2006; Pankonin et al., 2009). In the brain, neuregulin is concentrated in white matter astrocytes, and is elevated in the cerebrospinal fluid of AD patients (Pankonin et al., 2009), implying that it may be a response to ongoing myelin injury. The therapeutic inhibition of ß-secretase may thus produce the undesired effect of inhibiting myelin repair even as it helps reduce Aß formation. Therefore, if myelin repair is crucial to avert the onset of AD, the use of ß-secretase inhibitors may produce more harm than good by inhibiting neuregulin when it is most needed (Glabe, 2006; Figure 14.2). Furthermore, the control of acquired factors that damage white matter, to be considered later, could by itself reduce the generation of Aß because the brain has less need for the activation of neuregulin by ß-secretase.
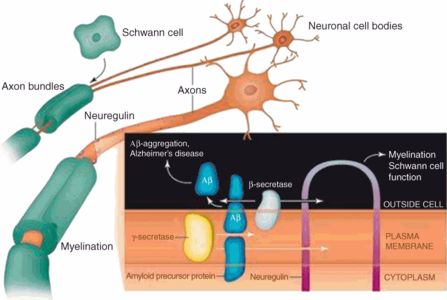
The dual action of ß-secretase: activation of neuregulin and generation of ß-amyloid (from Glabe, 2006).
These considerations naturally lead to a radically different approach to treatment of AD. Instead of striving to eradicate amyloid from the brain, clinicians may be well advised to recognize that this protein, and its downstream partner tau, can be seen as participating in an adaptive response to white matter injury, and efforts to prevent or repair this form of injury take precedence. The most effective treatment of AD may thus be the lifelong maintenance and protection of brain white matter. The eradication of amyloid may be useful in certain individuals at the point when the toxic effects of the protein begin to be clinically significant, but a more effective strategy could involve avoidance of white matter damage before any cortical protein deposition occurs. Both preventive and therapeutic strategies to achieve this objective can be imagined.
First, as the history of public health abundantly documents, prevention may be a most salutary intervention. Given the increasing recognition that AD is related to cardiovascular risk factors such as hypertension, diabetes, smoking, and obesity, and that TBI is another predisposing factor, opportunities for effective prevention are readily apparent (Qiu, Kivipelto, and von Strauss, 2009; Knopman and Roberts, 2010; Filley, 2015). In this regard, it has been proposed that AD should be considered a public health problem strongly related to preventable risk factors (Fotuhi, Hachinski, and Whitehouse, 2009). As these problems damage white matter before the appearance of the dementia of AD or even the signs of its precursor MCI, it is conceivable that early intervention using control of blood pressure and glucose, avoidance of tobacco, promotion of regular exercise and proper diet, and the wearing of seat belts and helmets may offer highly effective benefits with regard to preventing AD. As discussed in Chapter 13, a large European trial of hypertension control lasting about 4 years was able to show that dementia incidence was reduced by 55%, and, most important, this benefit was apparent not just for vascular dementia, but for AD as well (Forette et al., 2002). The fact that the only medication ever shown to lower the incidence of AD may exert a primary effect on white matter should serve as a stimulus for investigating this mechanism of action as a general preventive strategy.
Evidence for prevention of AD on a large scale has already begun to appear. Remarkably, several recent epidemiological studies from the United States and Europe have found a decline in the incidence and prevalence of dementia over the past two decades, and these statistics also include dementia related to AD (Larson, Yaffe, and Langa, 2013). These trends have been tentatively explained by the improving care of cardiovascular risk factors and access to education in industrialized nations (Larson, Yaffe, and Langa, 2013). One estimate holds that up to 50% of the worldwide burden of AD can be attributed to modifiable risk factors (Barnes and Yaffe, 2011). Thus the intervention of primary care and other physicians, combined with the institution of public health initiatives – all designed to address such factors as hypertension, diabetes, smoking, obesity, hyperlipidemia, physical inactivity, TBI, depression, sleep disorders, and cognitive inactivity – may bring about effective preventive measures for many cases of AD (Filley, 2015). Moreover, if the myelin model of AD is correct, other acquired insults to white matter may be identified and present additional opportunities for prevention.
As for treatment, it is assuredly the case that some people will still develop AD despite the best preventive efforts of medicine and society. In the context of the myelin model, this unfortunate event may occur for many reasons, including unavoidable medical disorders, accidental TBI, or a genetic predisposition in amyloid genes leading to excessive cortical amyloid deposition (Hardy, 2009). A significant component of genetic risk is also known in AD, clearly in early-onset disease (Howard and Filley, 2009), and to some extent in late-onset AD as well, with susceptibility conferred by the APOE ε4 allele, and other genetic effects that are being investigated with genome-wide association studies (Tosto and Reitz, 2013). These factors cannot be ignored, and it may be that therapies targeting amyloid or even tau may find a place in the therapeutic armamentarium for AD. Indeed, ongoing clinical trials with solaneuzumab and other similar antibodies may yet find success and offer solid support for the amyloid hypothesis.
Yet even when cortical proteins have accumulated and dementia has developed, attempts to enhance the endogenous repair capacities of the brain may still be effective. If damage to white matter precedes the appearance of plaques and tangles, this injury may continue throughout the disease, and efforts to repair white matter may be worth considering at any point in the disease course. In this regard, intriguing possibilities are raised by promising experimental results with the use of APOE-mimetic peptides (Sarantseva et al., 2009; Vitek et al., 2012) and neuregulin (Min et al., 2011; Woo et al., 2012), both of which may function to help repair white matter at an early stage of cognitive impairment, and prevent the excess deposition of amyloid that could lead to a less reversible state of cortical degeneration.
The myelin model of AD is of course hypothetical, and much remains to be done so that it may be confirmed or refuted. As the disease remains alarming because of its high prevalence and incurability, however, a new paradigm is eminently reasonable to consider. If the myelin hypothesis is correct, it may provide the long-sought answer to the scourge of AD, and the present goal of eradicating amyloid from the brain will be seen as a worthy but inadequate attempt to solve the problem.
References
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


