Axial fluid-attenuated inversion recovery (FLAIR) MRI scan of an older person with leukoaraiosis. Patchy white matter hyperintensties are scattered throughout the white matter.
The concept of LA was introduced by Hachinski and colleagues to designate the white matter “rarefaction” frequently seen on CT and especially MRI scans of older individuals with or without symptoms and signs of cerebral impairment (Hachinski, Potter, and Merskey, 1987). These changes appear as low-density white matter areas on CT and white matter hyperintensities on MRI, and the term “unidentified bright objects” was popular for several years in reference to the MRI changes (Román, 1996). The intent of the term LA was to provide a purely descriptive word for brain white matter abnormalities (Hachinski, Potter, and Merskey, 1987), which at that time were not well understood in terms of pathogenesis and clinical correlates. The caution embodied by the use of the term was indeed appropriate, as some quickly made the premature assumption that the changes of LA represented BD (Kinkel et al., 1985). With further work, however, both the origin and the significance of LA began to be understood, and the emerging information directly indicated a relationship to vascular compromise and BD.
As for pathogenesis, support for the ischemic origin of LA steadily accumulated (Pantoni and Garcia, 1997; Pantoni, Poggesi, and Inzitari, 2007; O’Sullivan, 2008). Neuropathological studies consistently found arteriosclerotic changes within areas of LA (Leifer, Buonanno, and Richardson, 1990; Fazekas et al., 1993). The small penetrating arterioles supplying the white matter were noted to manifest narrowing of the lumen secondary to the accumulation of hyaline material, and the sparing of subcortical U fibers further argued for selective small vessel involvement (Pantoni and Garcia, 1997). Gradually a consensus grew that LA reflects recurrent transient hypotension leading to incomplete infarction that damages the oligodendrocytes, myelin, and axons of cerebral white matter (Pantoni and Garcia, 1997; Román et al., 2002; O’Sullivan, 2008). Cerebral blood flow studies have supported this proposed pathogenesis by showing reduced white matter perfusion in areas of LA while gray matter is normally perfused (Markus et al., 2000). Studies of clinical populations found strong correlations between LA and cerebrovascular risk factors such as hypertension, diabetes mellitus, cardiovascular disease, and past history of stroke (Gerard and Weisberg, 1986; Inzitari et al., 1987) and, more recently, smoking (Fukuda and Kitani, 1996), obesity (Jagust et al., 2005), and the metabolic syndrome (Park et al., 2007). As helpful as these advances have proven, however, it must be remembered that LA remains a neuroradiological finding that may reflect physiologic changes such as dilation of perivascular Virchow-Robin spaces (état criblé) or periventricular caps and bands – all of which are benign – or other neuropathology such as the demyelinative plaques of multiple sclerosis (MS) (Merino and Hachinski, 2000; Barkhof and Scheltens, 2002).
As the ischemic origin of LA has been amply documented, and it has become clear that environmental factors play an important role in pathogenesis, a genetic contribution to LA has also been observed. A recent large European genome-wide association study (GWAS) involving more than 9,000 subjects identified a novel locus on chromosome 17 as associated with LA (Fornage et al., 2011). This discovery helps explain the considerable heritability of LA, reported as ranging from 55% to 80% (Fornage et al., 2011). As the first substantial evidence of a genetic influence on LA, these findings invite further investigation of what is becoming a complex pathogenesis. In this light, it is noteworthy that some evidence also supports an association of the apolipoprotein E ε4 allele with a higher burden of LA (Godin et al., 2009). Data now appearing therefore suggest that nature and nurture both contribute to the origin of LA.
The specific cerebral location of LA has attracted some attention, and evidence exists that the pathogenesis and sequelae of LA may differ depending on where it is found. From neuropathological studies of older people with incidental white matter hyperintensities, Fazekas and colleagues (1993) proposed that while subcortical lesions were ischemic, periventricular lesions were related to altered fluid dynamics producing white matter edema and subsequent demyelination. The functional consequences of lesions in these locations may differ as well, with subcortical lesions associated with cognitive dysfunction (Soumaré et al., 2009), and periventricular lesions associated with gait disorder (Blahak et al., 2009). Others have pointed out, however, that subcortical and periventricular lesions are highly correlated with each other, and thus a categorical distinction between them may be arbitrary; in this light, the clinical implications of subcortical versus periventricular lesions remain tentative (DeCarli et al., 2005).
The neurobehavioral significance of LA has come to be understood from many studies that have steadily yielded an ever more coherent account. Early investigations using low-field-strength MRI magnets and standard neuropsychological measures found no correlation of LA with cognitive dysfunction, suggesting that improved imaging and more specific cognitive measures might prove more useful (Rao et al., 1989c; Filley et al., 1989a). Subsequent research found that such correlations could indeed be detected, and the primary cognitive domains affected in LA were attention and cognitive speed (Junqué et al., 1990; van Swieten et al., 1991; Schmidt et al., 1993; Ylikoski et al., 1993). Large-scale MRI studies of older individuals began to appear, and correlations continued to be found between the severity of LA and cognitive dysfunction (Longstreth et al., 1996; de Groot et al., 2000; Au et al., 2006; Inzitari et al., 2007; Murray et al., 2010). More recently, longitudional study has documented that the advance of LA produces more severe cognitive dysfunction (Smith et al., 2015). With these impressive data sets, a consistent pattern has emerged of slowed processing speed and executive dysfunction, deficits found regardless of white matter lesion location, an observation that may be explained by the substantial convergence of numerous association tracts on the frontal lobes (Tullberg et al., 2004). To sum up a large body of work, comprehensive reviews have concluded that LA has been convincingly shown to have effects on cognition, with major effects on processing speed and executive function (O’Sullivan, 2008; Debette and Markus, 2010).
Most recently, newer neuroimaging techniques now in use have disclosed abnormalities in the normal-appearing white matter (NAWM) of people with LA. Perhaps not surprisingly, much of the brain in LA is affected beyond the extent of the obvious lesions seen on conventional MRI. Magnetic resonance spectroscopy (MRS) was found to reveal neurometabolite changes in the NAWM of individuals with LA consistent with myelin damage (Firbank, Minett, and O’Brien, 2003; Charlton et al., 2006). Diffusion tensor imaging (DTI) has been still more informative, with studies showing microstructural white matter abnormalities to correlate with impaired global cognition, processing speed, attention, working memory, and executive function (Jones et al., 1999; Charlton et al., 2006; Vernooij et al., 2009; van Norden et al., 2012). DTI changes have also been suggested to be more sensitive to longitudinal cognitive decline than the advance of LA on MRI (Charlton et al., 2010).
These studies on the NAWM in LA inform previous work on the idea of a threshold effect for cognitive decline. Boone and colleagues (1992) reported that a threshold of 10 cm2 of affected white matter on MRI was required before cognitive dysfunction could be detected. A 1993 consensus statement suggested that cognitive impairment occurred when LA involved 25% of the cerebral white matter (Román et al., 1993). Libon and colleagues (2008) extended this work by showing that only severe LA was associated with executive dysfunction. These studies recall similar observations in other white matter disorders that a certain threshold of disease burden is required before cognitive impairment occurs (Filley, 2012). The notion of a threshold effect is indeed plausible, but it is clear that LA seen on conventional MRI does not represent the entirety of ischemic neuropathology within the cerebral white matter. The NAWM data introduce considerably more complexity into this issue since the true extent of disease may be unclear. Moreover, as will be discussed, the impact of LA can also be mitigated by cognitive reserve, further confounding the interpretation of this finding.
It has also become clear that additional neurologic morbidity and mortality are associated with LA. Briley and colleagues (2000) found that LA predicts morbidity and mortality independent of previous neurologic deficits. An early MRI study found a significant association of LA with gait disorder (Longstreth et al., 1996), and longitudinal MRI has clearly documented progressive slowing of gait with the advance of LA (Smith et al., 2015). Smith (2010) summarized evidence that LA is important for determining stroke outcome as well as stroke incidence. Recent findings have also suggested that LA may increase the risk of anticoagulant-related hemorrhage in patients with atrial fibrillation or other conditions requiring anticoagulation (O’Sullivan, 2008). In view of the wide distribution of cerebral white matter, and the probability of multifocal white matter involvement interfering with the operations of multiple distributed neural networks, it should not be surprising that LA of sufficient magnitude can disrupt elemental as well as higher neurologic functions and compromise other important determinants of brain health.
Another intriguing aspect of the work on LA is that education appears to protect against the cognitive dysfunction produced by white matter lesions. A population-based study showed that an association between LA and cognitive dysfunction was present in individuals with lower education but not in more educated people (Dufouil, Alpérovitch, and Tzourio, 2003). These findings are similar to recent observations in other white matter disorders (Filley, 2012), and support the hypothesis that cognitive reserve conferred by education, which is plausibly mediated by increased cortical synaptic density, can mitigate the detrimental cognitive consequences of LA (Dufouil, Alpérovitch, and Tzouri, 2003).
The accumulated evidence now justifies the statement that LA is a largely ischemic phenomenon that predicts an increased risk of cognitive dysfunction, dementia, gait disorder, stroke, and mortality (Debette and Markus, 2010; Smith et al., 2015). The pattern of cognitive dysfunction in LA most typically implicates processing speed and executive function (O’Sullivan, 2008; Debette and Markus, 2010). These conclusions, combined with neuroradiological and neuropathological commonalities, lend support to the contention that LA lies on the same clinical-pathological spectrum as BD (Filley et al., 1988; van Swieten et al., 1991; Román, 1996; Libon et al., 2004; Debette and Markus, 2010). One important implication of this connection is that vigorous treatment of LA by modification of many well-recognized cerebrovascular risk factors may significantly impact the onset and manifestations of age-related cognitive decline. This and other implications of the relationship of LA to BD will now be taken up.
Binswanger’s Disease
In 1894, the Swiss neuropathologist Otto Binswanger sparked what became more than a century of controversy on white matter disease and dementia. In a three-part article on the differential diagnosis of general paresis of the insane, a common dementing disease of that time, Binswanger presented gross neuropathology from eight patients who had progressive dementia associated with marked white matter atrophy. The cortex was spared, but there was prominent atherosclerosis; he called this disease “encephalitis subcorticalis chronica progressiva” and related it to insufficient blood supply of the cerebral white matter (Blass, Hoyer, and Nitsch, 1991). Binswanger thus offered the seminal proposal that ischemic damage to white matter alone could lead to progressive cognitive decline.
Eight years later, Alois Alzheimer presented additional cases similar to those of Binswanger but with histologic observations supporting the idea that arteriosclerotic white matter disease could produce dementia (Alzheimer, 1902). It was in fact Alzheimer who linked the name Binswanger with this disorder (Román, 1987), thus establishing BD in the medical literature. Other names appeared, however, as time progressed. In 1962, Olszewski translated the articles of Binswanger and Alzheimer and presented two new cases emphasizing the importance of lacunar infarction, offering the alternative name “subcortical arteriosclerotic encephalopathy” (Olszewski, 1962). Today, many avoid the use of BD and prefer the more generic subcortical ischemic vascular dementia (SIVD) as a category of vascular dementia that includes both BD and the lacunar state (état lacunaire) of Pierre Marie (Román et al., 2002).
BD has been questioned as a clinical-pathological entity because Binswanger may not have been the first to describe the disorder, and because he provided no microscopic data in his reports to complement the gross neuropathologic findings (Hachinski, 1991). Olszewski (1962) even speculated that Binswanger’s cases could have had neurosyphilis. However, several authoritative reviews have endorsed the eponymic terminology (Babikian and Ropper, 1987; Fisher, 1989; Bennett et al., 1990; Caplan, 1995; Hurley et al., 2000; Román et al., 2002; Caplan and Gomes, 2010; Huisa and Rosenberg, 2014), and BD persists as a clinical entity in major neurology textbooks (Ropper, Samuels, and Klein, 2014). In this book, while the controversy about this disease will be acknowledged, the term BD will be used because Binswanger deserves credit for associating diffuse ischemic white matter disease with progressive dementia (O’Sullivan, 2008), and because the specific impact of BD on white matter renders the term most appropriate for our purposes.
BD can be considered a form of vascular dementia characterized by prominent involvement of the cerebral white matter. Its prevalence is not known because no definitive diagnostic test is available during life; although white matter lesions on neuroimaging scans of older people are suggestive, such lesions alone are insufficient for the diagnosis because they can be seen in many other diseases and in normal aging. Moreover, many neurologists are influenced by their corticocentric bias to make the diagnosis of coexisting Alzheimer’s Disease (AD) in patients with white matter lesions suggestive of BD. Despite these issues, BD may nevertheless be common. Nearly all older people have one or more ischemic white matter lesions on MRI (Román et al., 2002), and whereas most do not have BD, some autopsy studies have found that up to 35% of older dementia patients may have BD lesions at postmortem examination (Santamaria Ortiz and Knight, 1994). Evidence for BD existing as a dementia distinct from AD comes from study of patients with SIVD and severe white matter disease who also had Pittsburgh compound B (PiB) imaging with positron emission tomography to assess the burden of amyloid: More than two-thirds of these patients did not have cortical PiB retention (Lee et al., 2011), suggesting that BD may be more common than often thought. More satisfactory answers to the question of BD prevalence must await further study.
Clinically, BD is strongly associated with hypertension and other cerebrovascular risk factors, and presents in middle to late life as progressive neurologic and neurobehavioral dysfunction, sometimes, but not always, following a stepwise course (Babikian and Ropper, 1987; Fisher, 1989; Bennett et al., 1990; Caplan, 1995; Caplan and Gomes, 2010; Huisa and Rosenberg, 2014). Table 6.1 displays three approaches to the clinical diagnosis of BD. Neurologic features include focal corticospinal dysfunction, extrapyramidal signs, acute lacunar syndromes, gait disorder, and pseudobulbar affect. A useful sign of BD is diffuse hyperreflexia, a finding not typically present in AD patients with a comparable degree of dementia. Neurobehavioral manifestations include apathy, inertia, abulia, memory impairment, visuospatial dysfunction, depression, poor judgment, loss of insight, and relatively preserved language. The diagnosis may be difficult in early stages, when all of these features may be less apparent. Psychiatric dysfunction may develop before cognitive deterioration and neurologic signs (Lawrence and Hillam, 1995), a sequence typical of many other white matter disorders (Filley, 2012). Apathy and inertia can be mistaken for the cognitive slowing that is commonly seen in normal aging. Together with CT or MRI evidence of white matter vascular disease (Figure 6.2), however, BD can usually be diagnosed in life with considerable confidence (Bennett et al., 1990; Caplan, 1995; Huisa and Rosenberg, 2014). Whereas some clinicians hesitate to make this diagnosis because of the absence of accepted diagnostic criteria, and select instead SIVD or vascular dementia, inspection of Table 6.1 discloses that various sets of clinical and imaging criteria for BD actually have much in common.
| Bennett et al. (1990) | Caplan (1995) | Huisa and Rosenberg (2014) | |
|---|---|---|---|
| Clinical | Dementia on clinical examination and by neuropsychology Any two of the following: A vascular risk factor or evidence of systemic vascular disease Focal cerebrovascular disease Subcortical cerebral dysfunction (parkinsonian, magnetic, or senile gait; rigidity; gegenhalten; incontinence related to a spastic bladder) | Usual onset 55–75 years of age Men = women Acute strokes, often lacunar infarcts Subacute onset of focal signs Seizures during subacute progression Stepwise progression of motor, cognitive, and behavioral deficits Periods of stabilization, plateaus, and even improvement Pyramidal signs Extrapyramidal signs Abnormal gait Pseudobulbar signs Apathy, inertia, disinterest, abulia Poor judgment, lack of insight, altered affective responses Variable deficits in memory, language, and visuospatial function | Cognitive impairment Pyramidal signs Extrapyramidal signs Hypertension Ataxia, balance disturbance, falls Progressive symptoms |
| Imaging | Bilateral LA on CT, or Bilateral, multiple, or diffuse subcortical T2 lesions on MRI | Patchy, irregular PV WM attenuation Irregular focal PV and subcortical WM lesions Lesions in corona radiata and centrum semiovale, often large and confluent Multiple lacunar infarcts Hydrocephalus | WM hyperintensities Brain atrophy, mild to moderate Lacunar infarcts Microbleeds Enlarged perivascular spaces Intracranial atherosclerosis High pulsatility index by transcranial Doppler |
LA – leukoaraiosis, CT- computed tomography, PV– periventricular, WM – white matter, MRI – magnetic resonance imaging
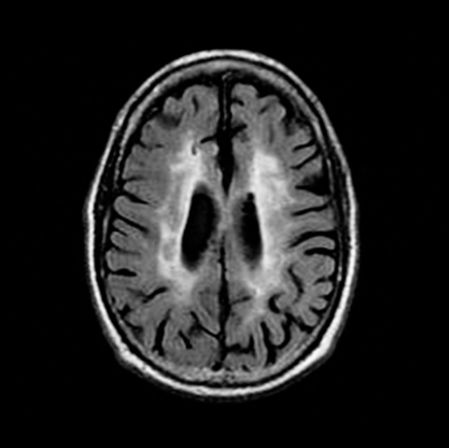
Axial fluid-attenuated inversion recovery (FLAIR) MRI scan of a patient with Binswanger’s Disease. Extensive periventricular hyperintensity is present.
Neuropathological observations constitute the basis for understanding the origin of dementia in BD. Hypertension is the most powerful risk factor, and the long penetrating arterioles of the deep cerebral white matter are invariably damaged by thickening and hyalinization of the vessel walls (Román et al., 2002). This arteriosclerosis in turn leads either to BD, in which hypoperfusion produces incomplete infarction of the white matter, or to the lacunar state, with occlusion of small vessels and completed infarcts leaving encephalomalacia (Román et al., 2002). The brain stem may be affected in BD (Pullicino et al., 1995), but the cortex is spared from the neuropathological process, and the subcortical gray matter is less affected than the white matter (Caplan, 1995; Caplan and Gomes, 2010). It has been shown in both human studies (Brown and Thore, 2011) and nonhuman animal studies (Pantoni, Garcia, and Gutierrez, 1996) that a high vulnerability of cerebral white matter to ischemia exists because of compromised perfusion from long penetrating arterioles superiorly and lenticulostriate arteries inferiorly. Microscopic findings early in the course of BD may be limited to myelin pallor, but in advanced cases there is loss of oligodendrocytes, myelin, and axons along with astrocytic gliosis; the subcortical U fibers are typically spared in BD, as they are in LA (Román et al., 2002). To summarize, BD can be seen to develop as complete and incomplete white matter infarctions accumulate (Román et al., 2002), sufficient to cause dementia from selective white matter injury and not because of comorbid neuropathology from AD (Lee et al., 2011).
Not surprisingly, the neuroradiology of BD is controversial because of unresolved questions about the nosologic status and diagnosis of the disease. CT initially provided some idea of lesion burden, and the use of MRI improved the identification of white matter changes to such an extent that many were led to conclude that these lesions establish the presence of BD (Kinkel et al., 1985). However, this view was soon abandoned as it was realized that white matter changes are not always associated with dementia. Currently, BD is a diagnosis suggested, but not confirmed, by the MRI white matter findings, and clinical correlation is essential. The ultimate decision about whether an older demented patient has BD can be made only at autopsy, and MRI is most useful in showing the type, location, and extent of white matter disease in vivo.
Recent advances in neuroimaging have led to more detailed studies designed to evaluate microvascular alterations within the white matter. As in LA, investigation has led to the recognition that the NAWM may not be normal in BD. MRS has documented microstructural changes in the NAWM of patients with vascular dementia (Jones and Waldman, 2004). Similarly, magnetization transfer imaging (MTI) of vascular dementia patients has shown reduced magnetization transfer ratio (MTR) most prominent within periventicular white matter lesions (Tanabe et al., 1999), and other investigators correlated decreased MTR with cognitive dysfunction in BD (Hanyu et al., 1999). DTI has also shown its value, disclosing that NAWM abnormalities in SIVD are more sensitive to early cognitive impairment than conventional MRI findings (Xu et al., 2010).
The characterization of cognitive dysfunction in BD has been clarified to some extent. Babikian and Ropper (1987) called attention to memory loss, confusion, apathy, and changes in mood and behavior that were typically not accompanied by aphasia, apraxia, and movement disorder. Román (1987) classified BD as a subcortical dementia because of the frequency of personality change, forgetfulness, and confusion, and the relative rarity of aphasia, apraxia, and agnosia. Stuss and Cummings (1990) endorsed this opinion, adding that the neurobehavioral profile of BD reflects frontal-subcortical dysfunction. Several clinical studies have also supported this characterization, with detailed mental status or neuropsychological examinations in BD patients documenting impaired executive function, attention, memory, visuospatial ability, and abstract thinking in the presence of relatively spared language, praxis, and gnosis (Kinkel et al., 1985; Sacquena et al., 1989; Lee et al., 1989; Caplan, 1995; Libon et al., 2004; Huisa and Rosenberg, 2014). Evidence thus exists to show that cognitive and emotional dysfunction in BD can be attributed to subcortical or frontal-subcortical pathology.
The specific contribution of the white matter disease to the dementia of BD is more difficult to establish, reflecting the generalization that white matter is more challenging to examine. But evidence has steadily accumulated. In early studies, lowered IQ scores were correlated with white matter lesions on CT scans (Loizou, Kendall, and Marshall, 1981), and correlations of cognitive decline with ischemic white matter changes were shown using MRI (Révész et al., 1989). Later, neuropsychological evaluation of BD patients found poor concentration, apathy, and cognitive slowing consistent with frontal lobe disturbance and white matter dysfunction (Bogucki et al., 1991). An important contribution came from study of stroke patients who were found to have limiting neurobehavioral dysfunction even after a single white matter lacunar infarct (Van Zandvoort et al., 1998). The gradual accumulation of incomplete infarctions within white matter in BD was found to have similar effects on cognition, as shown in a quantitative MRI study of vascular dementia patients that found a strong correlation between white matter lesion area and dementia (Liu et al., 1992). Further study of MRI white matter lesions led to the view that these lesions exert prominent effects on frontal-subcortical circuits and produce executive dysfunction (Desmond, 2002). More recent evidence supports a deficit profile in BD patients characterized by cognitive slowing, executive dysfunction, and impaired memory retrieval with sparing of language (Román et al., 2002; Libon et al., 2004; Huisa and Rosenberg, 2014).
Despite these advances, BD remains a controversial entity that serves to underscore several unresolved questions in the relationship between vascular white matter disease and dementia. Whereas neurologists have long understood the potential for cortical and subcortical gray matter infarcts to impact cognitive function, acceptance of the view that ischemic white matter lesions alone can produce dementia has at times been grudging. Clinicians can rightly point out that solitary white matter lacunes and even numerous MRI white matter hyperintensities may have no apparent cognitive correlates. In addition, the possibility that gray matter disease – cerebrovascular, degenerative, or both – exists in patients with numerous white matter hyperintensities produces continued reluctance to ascribe cognitive dysfunction to white matter lesions. However, more detailed study of cerebrovascular disease has disclosed important white matter–behavior relationships, and BD can be seen as a useful, albeit imperfect, example of how white matter disease can produce dementia (Filley et al., 1988; Filley, 1998, 2012).
Toxic Leukoencephalopathy
Neurotoxicology, the study of the effects of toxic agents on the nervous system, encompasses a wide and diverse range of toxins. Some exert their effects on the peripheral nervous system (PNS), and others damage the central nervous system (CNS), including the brain white matter. Many physical and chemical toxins have a predilection for producing white matter damage, and this selective toxic effect led to the recognition of a division of neurotoxicology called toxic leukoencephalopathy (Filley and Kleinschmidt-DeMasters, 2001). MRI has been central to the discovery and characterization of many of these intoxications, often enabling detection of subtle white matter involvement that was previously unappreciated. Four categories of toxic leukoencephalopathy can be distinguished: drugs of abuse, cranial irradiation, therapeutic drugs, and environmental toxins (Filley and Kleinschmidt-DeMasters, 2001). Table 6.2 lists the best-known white matter toxins within these categories. In this section, toluene abuse, radiation, and cancer chemotherapy will be discussed as particularly illustrative examples of how toxic injury illuminates the function of normal and abnormal white matter.
| Drugs of abuse Toluene Alcohol Cocaine Heroin Cocaine MDMA (3,4-methylenedioxy-methamphetamine) Psilocybin |
| Radiation |
| Therapeutic drugs Cancer chemotherapeutic agents Cyclosporine Tacrolimus Amphotericin B Hexachlorophene |
| Environmental toxins Carbon monoxide Arsenic Carbon tetrachloride |
Toluene leukoencephalopathy
Drugs of abuse are well known to cause injury to the nervous system, but the characterization of these intoxications has been been difficult because drug abusers are often exposed to more than one agent, and there exists a relative paucity of neuropathological studies of individuals with single exposures. As a general rule, brain injury from drugs of abuse features a wide spectrum of neuropathology, including ischemia, cerebrovascular disease, and a range of neuronal changes implicating additional inflammatory and degenerative mechanisms (Büttner, 2011). Specific white matter changes can be seen that suggest a direct leukotoxic effect, although much remains to be clarified (Büttner, 2011). The discussion to follow will consider a commonly abused drug for which substantial neuroimaging evidence, and in some cases neuropathological evidence, exists to document selective effects on white matter with neurobehavioral sequelae.
One of the more intriguing discoveries made possible by the application of MRI is toluene leukoencephalopathy, which convincingly illustrates the capacity for pure white matter damage to produce dementia (Hormes, Filley, and Rosenberg, 1986; Rosenberg et al., 1988a; Rosenberg et al., 1988b; Filley, Heaton, and Rosenberg, 1990). Toluene (methylbenzene) is a common household and industrial solvent that is also popular as a drug of abuse among millions of people in the United States and in many countries around the world. Inhalant abuse is a highly prevalent but underappreciated form of drug abuse; it is estimated that 9% of the United States population has experimented with inhalants, and that as many as 50% of these individuals are at risk of abuse (Filley, 2013). The abuse of toluene is practiced by the inhalation of solvent vapors derived mainly from spray paint, which leads to rapid inebriation and euphoria without a notable withdrawal state (Filley, Halliday, and Kleinschmidt-DeMasters, 2004). The solvent may be inhaled to achieve this effect on a daily basis for years without respite. If exposure is heavy and prolonged, a dramatic neurologic syndrome appears in which dementia is the most prominent component of a clinical proflile that also includes ataxia, corticospinal dysfunction, and various brain stem and cranial nerve abnormalities (Hormes, Filley, and Rosenberg, 1986). These effects may be persistent in many abusers even after abstinence is achieved, and the pattern of dementia in these individuals resembles that described in subcortical dementia (Hormes, Filley, and Rosenberg, 1986) and, more specifically, the profile of WMD (Filley, Rosenberg, and Heaton, 1990; Filley, Halliday, and Kleinschnidt-DeMasters, 2004). MRI scans of toluene abusers display diffuse T2 hyperintensity in the cerebral and cerebellar white matter, and the degree of cerebral involvement strongly correlates with the severity of dementia, the most prominent clinical manifestation (Filley, Heaton, and Rosenberg, 1990). Autopsy studies confirm the MRI findings by revealing selective myelin loss with sparing of the cerebral cortex, neuronal cell bodies, and even axons in all but the most severe cases (Rosenberg et al., 1988b; Kornfeld et al., 1994; Fornazzari et al., 2003; Filley, Halliday, and Kleinschmidt-DeMasters, 2004). A recent review of 30 studies of toluene misuse concluded that white matter is indeed the likely target of toluene, and that observed cognitive deficits are consistent with white matter damage (Yücel et al., 2008). Toluene leukoencephalopathy thus serves as a model of the toxic white matter disorders, and as a convincing example of how white matter disease can lead to dementia.
Early MRI studies of toluene abuse investigating the pathogenesis of dementia proved invaluable in documenting diffuse leukoencephalopathy in the cerebrum and cerebellum. Several findings were revealed, including diffusely increased periventricular white matter signal on T2-weighted images, loss of differentiation between the gray and white matter, and diffuse cerebral atrophy (Rosenberg et al., 1988b), and these observations were amply confirmed (Caldemeyer et al., 1993; Xiong et al., 1993; Yamanouchi et al., 1997). Some cases have shown T2 hypointensities in the thalamus and basal ganglia, which were initially attributed to iron deposition but later attributed to the partitioning of toluene in lipids within these areas (Unger et al., 1994). The leukotoxic predilection of toluene is also consistent with documented neuropsychological defcitis in processing speed, sustained attention, memory retrieval, and executive function (Yücel et al., 2008). Preliminary DTI studies have found that microstructural white matter abnormalities can also be detected in inhalant abusers (Yücel et al., 2010).
Neuropathological investigation of toluene leukoencephalopathy has established the selectivity of white matter involvement. Initial autopsy studies consistently disclosed widespread white matter changes in the brain that spared cortical and subcortical gray matter as well as axons (Rosenberg et al., 1988b; Kornfeld et al., 1994). Whereas true demyelination was not observed, an increase in very long-chain fatty acids in the cerebral white matter suggested a neuropathological commonality with adrenoleukodystrophy (Kornfeld et al., 1994). More recently, further evidence was presented from a large neuropathological study that discovered selective white matter damage in 22 of 75 solvent abusers, with sparing of gray matter and axons within the injured white matter; the 22 affected brains were from people who had undergone longer periods of abuse (Al-Hajri and Del Bigio, 2010).
The neurobehavioral sequelae of extended toluene abuse are clear, but the impact of low-level occupational exposure to toluene and other solvents remains uncertain (Filley, Halliday, and Kleinschmidt-DeMasters, 2004; Filley, 2013). Exposure to toluene and similar solvents is common in the household, and there is little reason for concerns about leukotoxicity in this setting, Workers exposed to solvents in industrial settings, however, often have a variety of neurobehavioral complaints that could be a result of exposure, but their symptoms, which typically include fatigue, poor concentration, memory loss, depression, and sleep disturbance, are nonspecific and frequently unaccompanied by neurologic findings or evidence of neuropsychological dysfunction. Moreover, determining a cause-and-effect relationship in this situation is difficult because many individuals are exposed to multiple solvents, suffer depression or anxiety, struggle with concurrent alcohol or other drug issues, or are involved in often protracted litigation. The issue has been controversial since the 1970s, when reports of the “chronic painters’ syndrome” began to appear from Scandinavia (Arlien-Søborg et al., 1979). Since then, many authoritiies have addressed this condition, also called chronic toxic encephalopathy and the psychoorganic syndrome, and opinions on its existence range from expressions of support (White and Feldman, 1987; Baker, 1994) to skepticism (Rosenberg, 1995; Albers et al., 2000).
At present, low-level exposure to organic solvents including toluene in the workplace cannot be regarded as hazardous to white matter. Neuropsychological testing offers a sensitive approach to the detection of deficits, but whether observed deficits are specific for leukotoxicity is often unknown (Rosenberg, 1995; Albers et al., 2000). Neuroimaging with CT has not been helpful, as CT studies of solvent-exposed workers typically fail to show cerebral atrophy (Treibig and Lang, 1993), and white matter is not well seen with this technique. One MRI study was able to show diffuse white matter hyperintensity in individuals exposed to industrial solvents when compared to age-matched control subjects (Thuomas et al., 1996), but many similarly exposed individuals have normal MRI (Rosenberg, 1995). Perhaps most convincing is an autopsy study of 98 individuals who had been exposed to organic solvents in the workplace; results showed no difference in brain weight compared to control brains and no specific neuropathology that could be attributable to solvent intoxication (Klinken and Arlien-Søborg, 1993). It thus remains true that accurate diagnosis of individuals in this setting is not straightforward, and many cases of alleged cognitive impairment after occupational solvent exposure remain unconvincing after detailed neurobehavioral evaluation. Whereas the leukoencephalopathy of toluene abuse remains the best example of solvent-induced neurobehavioral dysfunction and one of the most instructive types of white matter dementia, similar but less severe effects from low-level exposure to toluene or other solvents remain to be substantiated. Prospective, controlled studies will be necessary to establish whether toxicity occurs in this setting, the threshold of exposure above which the toxicity can be expected, and whether white matter injury is significant.
Radiation
Radiation delivered to the brain as a therapeutic modality for neoplasia is well established in the treatment of many primary and metastatic tumors. As with other modalities for treating cancer, however, radiation confers a substantial potential for toxicity. In the brain, this neurotoxic effect was formerly assumed to fall most heavily on the hippocampus, but recent years have witnessed increasing recognition of the leukotoxic effects of radiation. Thus the problem of radiation leukoencephalopathy has come to be appreciated as one of the major limitations of cranial irradiation (Dietrich et al., 2008; Perry and Schmidt, 2006; Greene-Schloesser and Robbins, 2012).
This idea of a leukotoxic effect of radiation first appeared more than three decades ago with the work of Sheline and colleagues, which established that three types of radiation injury can occur in the brain, all of which primarily affect the cerebral white matter (Sheline, Wara, and Smith, 1980). First, an acute reaction may occur during treatment and is characterized by a confusional state or a worsening of preexisting neurologic signs. This mild syndrome is typically self-limited, and is thought to result from cerebral white matter edema. Next comes the early delayed reaction, which develops as a so-called somnolence syndrome weeks to months after irradiation. This syndrome has been ascribed to cerebral demyelination, and slow recovery takes place after the cessation of radiation. The most severe radiation-induced white matter injury is the late delayed reaction, which appears months to years after therapy and presents as a progressive, often-fatal dementia resulting from widespread demyelination and necrosis. As the acute reaction typically subsides spontaneously, most information on radiation leukoencephalopathy has been gathered from study of the early and late delayed effects.
The most prominent clinical sequelae of any form of radiation leukoencephalopathy are neurobehavioral. In adults, mental status disruptions such as confusion, personality change, apathy, memory loss, and dementia have been often noted (Vigliani et al., 1999; Filley and Kleinschmidt-DeMasters, 2001; Greene-Schloesser and Robbins, 2012), and focal neurobehavioral signs may appear in association with localized neuroradiological abnormalities (Valk and Dillon, 1991). Learning disabilities have been described in children (Constine et al., 1988), and those under 5 years of age may fare worse than older children (Fletcher and Copeland, 1988). The incidence of cognitive impairment after radiation for brain tumors has been reported to be a high as 50% to 90% (Greene-Schloesser and Robbins, 2012), but this figure likely depends to a large extent on age, cumulative dose, concomitant chemotherapy, and comorbid vascular risk factors such as diabetes mellitus (Dietrich et al., 2008). Patients have been described with radiation-induced dementia and prominent white matter neuropathology on neuroimaging, cerebral biopsy, or autopsy; qualitatively, the dementia was similar to that seen with white matter diseases such as BD and normal pressure hydrocephalus (DeAngelis, Delattre, and Posner, 1989; Vigliani et al., 1999; Omuro et al., 2005; Greene-Schloesser and Robbins, 2012). In patients irradiated for tumors located at the base of the skull, neurocognitive deficits correlated with total radiation dose, and the pattern of impairments in cognitive speed, visuospatial skills, and executive function was consistent with injury to the subcortical white matter (Meyers et al., 2000). Learning and memory are also impaired, but, importantly, memory retrieval is affected more than encoding, suggesting primary dysfunction within frontotemporal white matter networks (Dietrich et al., 2008). Long-term follow-up study of patients with glioma treated with radiation has detected deficits in executive function and processing speed in association with white matter lesions (Douw et al., 2009).
The radiation dose thought to induce radiation leukoencephalopathy in adults has generally been reported as greater than 50 Gy, and children are considered vulnerable at lower doses (Schultheiss et al., 1995; Perry and Schmidt, 2006). The safe lower limit of brain irradiation is unknown, although a study of healthy adults who received a dose of 1.2 Gy demonstrated no decrement in attention, a domain quite sensitive to radiation effects (Wenz et al., 1999). Focal irradiation appears to have less neurobehavioral impact than whole brain irradiation (Taphoorn et al., 1994). Significant damage may still occur with focal irradiation, as was found in patients who received focal radiation for nasopharyngeal carcinoma and developed prominent memory and language deficits in association with bilateral temporal lobe white matter necrosis (Cheung et al., 2000).
Evidence acquired since the initial studies of Sheline and colleagues (1980) has confirmed that the cerebral white matter is the major site of radiation injury to the brain (Perry and Schmidt, 2006). Neuropathological abnormalities in radiation leukoencephalopathy may be diffuse or focal, depending on the site(s) of irradiation. In general, a spectrum of changes reflects the range of severity that can develop – from edema to demyelination and ultimately necrosis (Vigliani et al., 1999; Filley and Kleinschmidt-DeMasters, 2001). Radiation does not produce significant damage to the cerebral cortex, and the often-diagnosed cortical atrophy in irradiated patients more likely reflects diminution of white matter volume (Valk and Dillon, 1991; Rogers et al., 2011). Hypothesized causes of radiation leukoencephalopathy include direct oligodendrocyte injury with secondary disturbance in myelin metabolism, damage to vascular endothelium resulting in breakdown of the blood–brain barrier and subsequent edema and demyelination, neuroinflammation, and oxidative stress (Sheline, Wara, and Smith, 1980; Vigliani et al., 1999; Greene-Schloesser and Robbins, 2012). Recent studies have underscored the potential importance of damage to oligodendrocyte progenitor cells (Dietrich et al., 2008).
Neuroimaging studies reflect the spectrum of neuropathological injury from radiation, and MRI findings become more severe with early delayed and particularly late delayed injury. MRI studies have supported an association between greater cognitive impairment and more extensive radiation-induced white matter disease (Corn et al., 1994; Armstrong et al., 1995; Mulhern et al., 1999; Schuitema et al., 2013). A study of medulloblastoma survivors demonstrated a direct correlation between decreased white matter volume due to radiation and a lower mean intelligence quotient (Mulhern et al., 1999). Armstrong and colleagues conducted longitudinal studies of the effects of radiotherapy, and found a decline in cognitive function between 1.5 and 4.5 months after radiation followed first by improvement and then later by a decline again at 2 years; these results were thought to be consistent with the time course of early delayed and late delayed radiation leukoencephalopathy (Armstrong et al., 1995). These investigators also provided evidence that memory retrieval deficits were particularly prominent and may represent a sensitive clinical marker of white matter neuropathology (Armstrong et al., 1995; Armstrong et al., 2000; Armstrong, Stern, and Corn, 2001).
Cancer chemotherapy
Many drugs used for cancer treatment may produce leukoencephalopathy that is clinically, neuropathologically, and neuroradiologically similar to that produced by radiation (Perry and Schmidt, 2006; Deitrich et al., 2008). The clinical effects of these drugs closely resemble those of radiation leukoencephalopathy, and reflect disruption of neurobehavioral function in a similar pattern, with lassitude, drowsiness, confusion, memory loss, and dementia (Lee, Nauert, and Glass, 1986; Moore-Maxwell, Datto, and Hulette, 2004). In clinical practice, radiation and chemotherapy are often administered together, so the toxic effects on the brain are compounded (Perry and Schmidt, 2006). Similarly, the neuroimaging appearance of cancer drug neurotoxicity can closely mimic that of cerebral radiation. Combined treatment typically produces more severe leukoencephalopathy, particularly if the chemotherapy is given by the intrathecal or intraventricular routes (Lee, Nauert, and Glass, 1986; Perry and Schmidt, 2006). However, the capacity of chemotherapeutic drugs alone to be specifically toxic to myelin has been emphasized (Deitrich et al., 2008; Meyers, 2008).
A particularly fulminant form of chemotherapy-related white matter injury is diffuse necrotizing leukoencephalopathy, in which progressive dementia leading to death is thought to result from axonopathy in addition to myelin damage (Rubinstein et al., 1975; Perry and Schmidt, 2006). This unusual disorder illustrates the general principle that axonal loss typically worsens the prognosis after white matter damage of any kind is sustained (Medana and Esiri, 2003).
The first antineoplastic drug recognized to produce leukoencephalopathy was methotrexate, which can be associated with the syndrome when given intrathecally or intravenously (Gilbert, 1998) or even orally in exceptional cases (Worthley and McNeil, 1995). High-dose intravenous methotrexate may cause leukoencephalopathy that manifests clinically as personality change, progressive dementia, and stupor (Allen et al., 1980). Another agent, BCNU (1,3-bis(2-chloroethyl)-1-nitrosourea), may also cause drug-induced leukoencephalopathy, whether given intravenously (Burger et al., 1981) or intra-arterially (Kleinschmidt-DeMasters and Geier, 1989). Progessive dementia and a fatal outcome may ensue (Kleinschmidt-DeMasters and Geier, 1989).
A number of other antineoplastic drugs may produce this syndrome. Cytosine arabinoside, 5-fluorouracil, levamisole, fludarabine, cisplatin, thiotepa, interleukin-2, and interferon-alpha are on this list (Filley and Kleinschmidt-DeMasters, 2001), and more can be expected. In general, these drugs share similar clinical features of toxic leukoencephalopathy, and their effects are typically well seen on MRI scans. The neuropathology of these intoxications, when available, documents variable degrees of cerebral demyelination and necrosis. Many cases show some reversibility, but more intense exposure seems to be associated with more severe leukoencephalopathy and a worse prognosis (Filley and Kleinschmidt-DeMasters, 2001). As with radiation, recent studies have suggested that damage to oligodendrocyte progenitor cells may be involved in more severe cases (Deitrich et al., 2008).
Traumatic disorders
Trauma to the brain can occur because of accidents, falls, assaults, military combat, sporting contests, or therapeutic interventions (Box 6.2). By far the most important form of brain trauma is the category known as traumatic brain injury (TBI), which includes closed or penetrating head injuries that impact brain structure and function among individuals engaged in many civilian activities and in military combat. One of the most urgent problems confronting medicine and society, TBI constitutes one of the most prevalent neurologic disorders. The understanding of neuropathological changes in the brain caused by physical trauma, and how they affect clinical presentation and recovery, is challenging because of the multiple adverse consequences of trauma on brain integrity, and there is no single neuropathological lesion of TBI. Whereas gray matter regions of the brain can be affected by trauma, it is clear that the white matter is significantly damaged, and approaching TBI from this perspective offers instructive new insights into the role of white matter in cognition more generally.
Traumatic brain injury
After a long period of relative neglect (Goldstein, 1990), TBI has recently been the object of considerable and well-deserved interest in neurology. TBI is an important source of neurobehavioral disability and death, estimated to affect 3.6 million Americans per year (Coronado et al., 2011), and serves as the primary example of traumatic white matter damage. Severe TBI may of course be an immediately fatal event, and in the United States more than 50,000 indivduals perish from TBI each year (Coronado et al., 2011). Many more individuals survive, and although most concussions resolve fully within weeks or a few months, it is sobering to consider that more than 5 million Americans are living with the chronic neurologic sequelae of TBI (Chauhan, 2014). TBI is especially problematic because of its high incidence in young adulthood, often leaving patients with decades of disability and lost productivity. The recent military conflicts in Iraq and Afghanistan have accentuated the problem, as many combatants return from active duty with blast injury, a new form of TBI distinct from blunt physical impact. Whatever the mechanism of injury, deficits in cognition and emotional status are typically the most problematic after TBI, far outpacing physical disability. The substantial initial recovery of physical function often misleads clinicians and families to anticipate a good neurobehavioral outcome that may never occur.
The terminology of neurobehavioral impairment in TBI has traditionally been separated from that of dementia, as many prefer to use “dementia” in reference mainly to older people with degenerative diseases. However, the presence of long-standing deficits in multiple cognitive domains in moderate to severe TBI patients surely justifies the descriptor “dementia,” and given the prominence of white matter neuropathology with head injury, TBI fully merits inclusion in this book. The dementia of TBI can also be referred to as static encephalopathy, another reasonable categorization, but however this condition is described, the lasting cognitive syndrome produced by TBI is part of a multifaceted illness that prohibits usual social and occupational function (Masel and DeWitt, 2010). Our goal at this point is to highlight the role of white matter injury in the pathogenesis of this lifelong disorder. A related and rapidly emerging topic – the relationship of TBI to degenerative dementia – will be considered in Chapters 14 and 15.
The clinical presentation of TBI necessarily includes impairment of mental status. In all cases, TBI involves some compromise of neurobehavioral function, ranging from transient loss of consciousness, confusion, or amnesia from concussion (Kelly et al., 1991) to the vegetative state following severe injury (Adams, Graham, and Jennett, 2000). The immediate severity of TBI is most often classified on the basis of initial Glasgow Coma Scale (GCS) score (Teasdale and Jennett, 1974) as mild (13–15), moderate (9–12), or severe (3–8), but this measurement is frequently unavailable. Most often, clinicians are faced with the task of diagnosing TBI in retrospect, which can be very challenging unless the injury is obvious. Similarly, the neuropathological basis of neurobehavioral impairment in TBI is often unclear. Despite these uncertainties, however, evidence developed over the past half century has steadily supported the notion that the primary origin of neurobehavioral sequelae after TBI is damage to the cerebral white matter.
Among the many lesions head trauma may produce, including cortical contusion, intracerebral hemorrhage, subdural hematoma, epidural hematoma, penetrating wounds, and hypoxic-ischemic damage, the most important TBI lesion is diffuse axonal injury (DAI) within the white matter (Strich, 1970; Adams et al., 1982; Gennarelli et al., 1982; Alexander, 1995; Adams, Graham, and Jennett, 2000; Smith, Meaney, and Shull, 2003; Chauhan, 2014). Also known as traumatic axonal injury (TAI), DAI produces widespread areas of white matter damage (Adams et al., 1982; Alexander, 1995; Kraus et al., 2007), and has been termed the signature neuropathology of TBI (Chauhan, 2014). Figure 6.3 shows a coronal brain section from a young man with severe, fatal TBI who sustained widespread DAI.
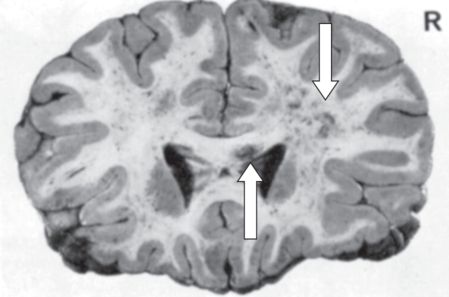
Diffuse axonal injury on postmortem coronal brain section of a young man with severe traumatic brain injury (from Strich, 1970). Both cerebral hemispheres are involved, the right more than the left (downward arrow), and the corpus callosum is also injured (upward arrow).
A key insight in this field is that DAI is present in all cases of TBI, from the mildest to the most devastating (Alexander, 1995). This lesion is responsible not only for the acute neurobehavioral effects of TBI, but also for chronic sequelae, including persistent attentional, executive, comportmental, and memory disturbances (Alexander, 1995), and even the vegetative state (Adams, Graham, and Jennett, 2000). Damage to frontal lobe white matter is particularly detrimental, compromising long-term outcome in many patients by interfering with restoration of normal personality, occupational function, and community reintegration (Filley, 2011).
Clinical and experimental studies of TBI have long supported the prominence of injury to the white matter (Strich, 1956; Adams et al., 1982; Gennarelli et al., 1982; Büki and Povlishock, 2006; Chauhan, 2014). An early term for this lesion in patients with severe posttraumatic dementia was “diffuse degeneration of the cerebral white matter” (Strich, 1956), and later the more specific descriptor “shearing injury” was introduced (Strich, 1961). Subsequently, variable degrees of DAI were demonstrated in both mild TBI (Oppenheimer, 1968) and severe TBI (Adams et al., 1982). The work of Nevin (1967) showed that white matter pathology was present in all individuals who survived more than a week after severe TBI, and the identical pattern of DAI was shown in experimental animals subjected to TBI (Gennarelli et al., 1982). The familiar term DAI was proposed to designate widespread injury to axons within the white matter of the injured brain (Adams et al., 1982), and TAI was used to designate the same process (Büki and Povlishock, 2006). Whereas these terms both point to brain axons as the primary sites of injury, it has now beome clear that myelin is concomittantly damaged (Chauhan, 2014; Mierzwa et al., 2015; Armstrong et al., 2015), and thus DAI serves to designate the white matter damage of TBI that is capable of producing major neurobehavioral sequelae.
Microscopically, DAI was first characterized by the presence of axonal retraction balls, microglial clusters, and Wallerian degeneration in white matter tracts (Gennarelli et al., 1982), and myelin injury is now also recognized (Chauhan, 2014; Mierzwa et al., 2015; Armstrong et al., 2015). Areas most affected by DAI are the dorsal midbrain, the corpus callosum, and the hemispheric white matter (Filley, 2011). The pathophysiology of DAI centers on shearing forces generated in the brain by sudden acceleration and deceleration (Adams et al., 1982; Gennarelli et al., 1982; Alexander, 1995; Büki and Povlishock, 2006; Chauhan, 2014). Rotational forces appear to be most deleterious, often producing instant loss of consciousness by shearing injury of white matter in the brain stem. Long association and commissural fiber tracts are also affected, as they are highly vulnerable to mechanical disruption. Injury to blood vessels producing multiple hemorrhagic foci within the white matter is also common in TBI. Further investigation has elucidated a cascade of cellular events occurring after TBI, including calcium entry into damaged axons, that contribute to additional axonal and myelin disruption (Büki and Povlishock, 2006). The extent of DAI correlates with clinical measures of severity, including the GCS, the length of unconsciousness, and the duration of posttraumatic amnesia (Alexander, 1995; Adams et al., 2011). Taken together, these observations serve to identify the essential difference between mild and more severe forms of TBI as the degree of DAI.
Neuroimaging studies have generally supported neuropathological findings emphasizing the importance of white matter damage in TBI. Most patients with mild TBI have normal conventional MRI, reflecting the microscopic nature of DAI. Early reports using CT in TBI patients demonstrated small focal hemorrhages in the white matter (Zimmerman, Bilaniuk, and Gennarelli, 1978), but CT has since been acknowledged as generally insensitive to the lesions of DAI (Kim and Gean, 2011). The improved sensitivity of MRI was demonstrated by observations that some brain-injured individuals with normal CT scans have white matter lesions on MRI (Mittl et al., 1994). An early prospective MRI study showed that DAI was the most common neuroradiological lesion in TBI, followed by cortical contusions, and that these lesions typically occur in the dorsal brain stem, corpus callosum, and hemispheric white matter, the same sites identified from neuropathological studies (Gentry, Godersky, and Thompson, 1988). With further technical advances, gradient echo (GRE) and susceptibility-weighted images (SWI) became available, and these sequences are now recommended to improve DAI detection because they reveal shearing-related microhemorrhages that accompany traumatic axonal and myelin injury (Kim and Gean, 2011).
The fact that microscopic lesions of DAI are often undetectable with conventional MRI explains why correlations between MRI white matter changes and neuropsychological function have often been modest (Levin et al., 1992). It remains true that one of the major issues with mild TBI is establishing the diagnosis in clinical practice without a sensitive and specific neuroimaging method. GRE and SWI sequences offer some help in this regard, but many DAI lesions still go undetected. As this problem was recognized, more sensitive MRI techniques were sought in the effort to improve white matter–behavior correlations, and the study of NAWM in TBI was initiated. Reduced white matter N-acetyl aspartate (NAA) on MRS was found to correlate with TBI severity (Garnett et al., 2000), and MTI detected white matter abnormalities that correlated with cognitive impairment (Bagley et al., 2000; McGowan et al., 2000). DTI showed similar results, notably in a study of children in whom a composite measure of white matter integrity was related to global outcome and processing speed after TBI (Levin et al., 2008). In longitudinal studies of adults with TBI, DTI disclosed DAI in multiple tracts, and acute abnormalities correlated with impaired learning and memory while lasting abnormalities correlated with slowed processing speed and executive dysfunction (Wang et al., 2011).
The specific neurobehavioral impact of DAI can be difficult to determine in many cases because other neurologic and systemic injuries in TBI also contribute to overall outcome. For example, patients with diffuse injury and superimposed cortical lesions fare worse than those with diffuse injury alone (Filley et al., 1987). However, available data are useful in developing a profile of neurobehavioral deficits that can be ascribed to the effects of DAI. In general, attention, memory, and executive function are most affected in TBI (Wilson and Wyper, 1992), and they also tend to dominate in mild TBI patients who go on to develop the postconcussion syndrome (Alexander, 1995). Sustained attention or concentration may be particularly affected, in contrast to simple attention (Kaufmann et al., 1993). Memory impairment has been associated with ventricular enlargement that is most likely a result of white matter volume loss (Anderson and Bigler, 1995). An intriguing observation is that TBI patients may display relative preservation of procedural memory compared to declarative memory (Ewert et al., 1989). More detailed examination of memory functions also reveals that this sparing of procedural memory may be accompanied by specific difficulty with memory retrieval (Timmerman and Brouwer, 1999). Executive dysfunction has been documented in TBI, and DTI studies have been used to relate this deficit to DAI within frontal lobe connections (Kinnunen et al., 2011). In contrast to these deficit areas, language is relatively preserved after TBI. An early report using Wechsler Adult Intelligence Scale (WAIS) verbal and performance IQ scores after TBI indicated that language is less affected and recovers more quickly than nonverbal skills (Mandleberg and Brooks, 1975). Personality and emotional changes, on the other hand, are frequent. Disinhibition or impaired impulse control may be disabling because of the social disruption that limits or precludes reintegration into society, and depression occurs in nearly half of patients with TBI (van Reekum, Cohen, and Wong, 2000). Cognitive slowing, an emerging feature of white matter dysfunction, has been associated with DAI using MRI volumetric analysis (Levine et al., 2006). A more recent DTI study found that decreased fractional anisotropy (FA) in several cerebral white matter tracts was associated with executive dysfunction among patients with moderate or severe TBI (Spitz et al., 2013). In sum, TBI leads to a wide range of neurobehavioral deficits, and the central role of white matter pathology is supported by the presence of DAI combined with a clinical profile matching that of other white matter disorders.
A discussion of this topic would not be complete without a review of blast injury. Military conflicts in the Middle East in the past decade have brought to attention this new form of TBI, which offers new challenges in terms of understanding pathogenesis and providing treatment (Ling et al., 2009). Many combatants in the Iraq and Afghanistan wars have been subjected to injuries related not to direct head trauma but rather to the impact of high-velocity air, smoke, gas, and debris from a nearby explosion. Symptoms after blast injury are very similar to those following more conventional concussion, leading to the notion that similar mechanisms of brain injury, including DAI, are involved, but the frequent co-occurrence of posttraumatic stress disorder has complicated analysis of these patients. To address this issue, DTI studies are beginning to document multifocal DAI as a mechanism of brain damage associated with blast injuries (Mac Donald et al., 2011; Hetherington et al., 2015).
The question now arises as to the risk of developing dementia after TBI as a result of DAI. There can be no definitive answer to this question, because the term dementia is not often used in the study of TBI outcomes, and the neuropathology of TBI includes but is not confined to DAI. Surely a substantial number of individuals with TBI, in whom DAI is a primary form of injury, must endure lasting intellectual dysfunction that meets criteria for dementia, but specific data on this issue are unavailable. However, some perspective can be achieved by considering the notion of disability, a term used in the TBI literature that serves as a rough equivalent of dementia and may be associated with DAI. The most widely employed measure of outcome after TBI is the Glasgow Outcome Scale (GOS; Jennett and Bond, 1975), which establishes the five outcomes of death, vegetative state, severe disability, moderate disability, and good recovery. TBI outcome data using the GOS, not surprisingly, are quite variable, as many factors influence recovery in any given patient, but lasting cognitive impairment precluding return to normal function has been documented in those who have experienced moderate or severe TBI (Sherer, Madison, and Hannay, 2000; Dikmen et al., 2009). Patients may show improvement over time, but even if they reach good recovery, residual cognitive effects usually preclude normal social and occupational function (Sherer, Madison, and Hannay, 2000). In patients studied neuropsychologically 2 years after moderate and severe TBI, cognitive performance is typically at the 20th percentile compared to matched controls (Schretlein and Shapiro, 2003), and return to work – which rarely assumes pre-injury levels – is possible for only about two-thirds of those with moderate TBI and one-third of those with severe TBI (Sherer, Madison, and Hannay, 2000). Whereas studies are needed to examine correlations between DAI and dementia, the importance of DAI in the pathogenesis of disability after TBI has been emphasized (Medana and Esiri, 2003).
A more recent issue with regard to dementia after TBI is the possibility of degenerative disease developing later in life. It may be that, in addition to the static encephalopathy produced by moderate or severe injury, progressive neurodegeneration may occur as a late sequel in some individuals who have sustained TBI. The two varieties of degenerative dementia most often discussed are AD and chronic traumatic encephalopathy (CTE), with current evidence suggesting an association of AD with moderate or severe TBI, and a link between CTE and multiple episodes of mild TBI (DeKosky, Ikonomovic, and Gandy, 2010). This topic warrants close scrutiny from the perspective of white matter neurobiology and will be considered in more detail near the end of this book. At present, the link between TBI and degenerative dementia – either AD or CTE – should be regarded as investigational, but increasing evidence suggests that each disease may result from the very common TBI lesion known as DAI.
Demyelinative diseases
This category of white matter disorders includes multiple sclerosis (MS) and many related but less common inflammatory diseases of myelin (Box 6.3). In terms of higher function, MS has recently been better appreciated as a source of cognitive and emotional impairment, recalling the initial insights of Jean-Martin Charcot in the 1870s (Charcot, 1877). A primary source of cognitive impairment is presumed to be white matter damage, as many studies have found at least modest correlations between extent of MRI white matter lesion burden and the degree of cognitive loss (Filley, 2012). More sophisticated MRI techniques have also documented abnormalities in the NAWM, indicating that subtle white matter pathology may be missed by conventional MRI. Recent studies, however, have also pointed toward cerebral cortical involvement, a form of the disease known as “cortical MS” (Stadelmann et al., 2008). This issue brings up the complexity of MS, and, like other white matter disorders, restriction of the neuropathology to white matter alone is uncommon. Nevertheless, much evidence supports the role of white matter involvement in the pathognesis of cognitive impairment and dementia in MS. This disease will now be discussed as the prototype demyelinative white matter disorder.
Multiple sclerosis
After more than a century of study, MS remains a perplexing disease that attracts the attention of neurologists and neuroscientists because of its clinical variability, uncertain etiology, and ever-improving therapy (Noseworthy, 1999; Hauser and Oksenberg, 2006). Among the many questions with MS requiring continued investigation are the characterization, significance, and treatment of neurobehavioral dysfunction. As recognized by Charcot (1877), both cognitive and emotional disturbances are apparent, but many details of these aspects of MS call out for a more thorough understanding. The wide range of neurobehavioral disturbances that compromise the life of individuals with MS presents a challenge to clinicians and an opportunity for researchers.
Cognitive impairment is an important clinical problem affecting many patients with MS (Rao, 1986; Feinstein, 2007; Langdon, 2011). This disturbance may range from subtle cognitive loss that can easily escape clinical detection to severe dementia that leaves no option except total care. Cognitive impairment can be a problem at all stages of the disease, potentially eroding physical independence, coping skills, driving, employment, medication compliance, and rehabilitation potential (Langdon, 2011). A generally recognized figure for the prevalence of cognitive impairment in MS is 40% to 70% (Langdon, 2011), and dementia, although less common, has been reported in up to 23% of MS patients (Boerner and Kapfhammer, 1999). For most of the history of MS, however, the high prevalence of cognitive impairment was not well appreciated. As late as 1970, for example, the prevalence of cognitive dysfunction of any kind in MS was estimated to be approximately 5% (Kurtzke, 1970). The use of more sensitive neuropsychological tests and improved research design in subsequent studies raised this figure much higher. Peyser and colleagues, for example, found a prevalence of 55% in their series of hospitalized MS patients (Peyser et al., 1980). Later, Heaton and colleagues considered the two major subtypes of relapsing-remitting and chronic-progressive MS, finding that 46% and 72%, respectively, were cognitively impaired (Heaton et al., 1985). A widely cited study of Rao and colleagues (1991) showed that cognitive disturbances are not confined to MS patients referred to university hospital clinics; using a community-based sample, they found a prevalence of 43%. A general consensus has long held that more severe forms of the disease predict more severe cognitive loss.
A key point is that cognitive impairment in MS may not be associated with more obvious features of neurologic disease. While memory and other disturbances may often appear together with elemental neurologic dysfunction in MS, cognitive loss may be isolated and by itself constitute the major source of disability. Clinicians working with MS patients are well advised to be aware of this possibility, as it is often overlooked in those who may be assumed cognitively normal because of the absence of motor and sensory findings on examination. Franklin and colleagues (1989) presented 12 patients with significant cognitive dysfunction, but whose physical disability as measured by the Extended Disability Status Scale (EDSS; Kurtzke, 1983) was minimal. These cases highlight the point that the EDSS, still widely used as a clinical measure of overall disability in MS, is generally insensitive to neurobehavioral dysfunction because of its emphasis on motor disability (Franklin et al., 1990).
Cognitive impairment can even appear as the sole feature of an MS exacerbation. Recent years have witnessed the recognition of the “isolated cognitive relapse” in MS, defined as a transient decline in neuropsychological performance in the absence of other neurologic involvement, and with gadolinium enhancement on brain MRI documenting inflammatory demyelination (Pardini et al., 2014). This newly recognized form of relapse, predictable from the natural history of the disease and now measurable with modern neuroimaging, contributes to gradual cognitive decline over time in patients with relapsing-remitting MS (Pardini et al., 2014).
Clinicians can also be misled by the often-subtle nature of cognitive impairment in MS compared to that of more familiar dementia syndromes such as that caused by AD. The typical pattern of cognitive deficits in AD, for example, features prominent amnesia, aphasia, apraxia, and agnosia, whereas MS patients are more likely to have difficulty with processing speed and sustained attention (Filley et al., 1989b; Rao et al., 1991). These distinctions imply that the use of routine cognitive screening methods may be inadequate. Because the dementia of MS, similar to that of many other white matter disorders, does not significantly disrupt language, heavily language-weighted measures such as the Mini-Mental State Examination (MMSE; Folstein, Folstein, and McHugh, 1975) are not well designed for the detection of cognitive loss (Franklin et al., 1988; Swirsky-Sacchetti et al., 1992).
In view of the inadequacy of both the EDSS and the MMSE for identifying cognitive dysfunction in MS, more detailed office testing or neuropsychological evaluation may be required to confirm a clinical suspicion of cognitive dysfunction. If time permits, a thorough mental status examination can often reveal the critical neurobehavioral deficits in MS patients. In other cases, referral for neuropsychological assessment is appropriate for the documentation of progressive cognitive decline, as in cases with incapacitating executive dysfunction who may otherwise have a benign clinical course (Filley, 2000). Newer approaches to this problem have also been developed by investigators seeking brief cognitive screening batteries. The Brief Repeatable Battery of Neuropsychological Tests for MS (BRB-MS; Rao et al., 1991), the Multiple Sclerosis Functional Composite (MSFC; et al., 1999), and the Minimal Assessment of Cognitive Function in MS (MACFIMS; Benedict et al., 2002) all offer sensitive techniques to detect cognitive impairment in MS.
Understanding the pathogenesis of cognitive impairment in MS begins with the neuropathology of the disease. Some patients have mainly spinal cord disease, but essentially all individuals with MS experience some degree of demyelinative plaque burden in the brain. The distribution of cerebral plaques in MS was studied by Brownell and Hughes (1962), who found that periventricular lesion sites were the most common, that the left and right hemispheres were equally affected, and that plaques were distributed proportionately throughout the white matter. A smaller number of plaques was also noted in cortical and subcortical gray matter regions (Brownell and Hughes, 1962). The classic pattern of white matter disease is well recognized on neuroimaging, with MRI studies of MS typically showing periventricular and callosal hyperintensities on T2-weighted and fluid-attenuated inversion recovery (FLAIR) images (Figure 6.4) consistent with prior neuropathological observations (Brownell and Hughes, 1962).
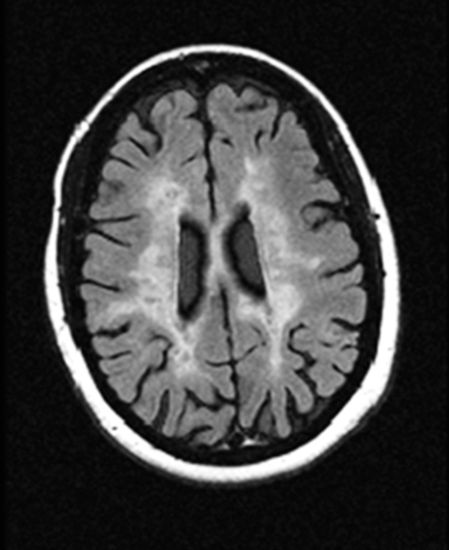
Axial fluid-attenuated inversion recovery (FLAIR) MRI scan of an individual with moderately advanced MS. Widespread subcortical demyelination is evident (courtesy of John R. Corboy, MD).
Neuropathological and neuroimaging advances have clarified the pathophysiology of white matter lesions in MS (Trapp et al., 1998; Simon, 2005). Considering MS as fundamentally an inflammatory disease, the first MRI sign of an acute MS lesion is an area of gadolinium enhancement within the white matter, optimally seen in axial T2-weighted or FLAIR images (Simon, 2005). The enhancement reflects inflammatory demyelination, which lasts for several weeks before it subsides and the lesion becomes either a T2 white matter hyperintensity or, with more extensive damage, a T1 “black hole” (Adams et al., 1999). As T2 hyperintensities reflect an increase in the water content of the affected white matter region, they are nonspecific and can result from other neuropathological processes. T1 lesions in MS, however, typically imply robust demyelination and axonal destruction that often occur in MS. An influential discovery in MS was the finding of axonal transection in many MS lesions, which reflects the intensity of inflammation in affected regions (Trapp et al., 1998). Axonal loss has in fact become widely recognized as a sign of a less favorable prognosis in patients with not only MS but many white matter disorders (Medana and Esiri, 2003). Axonal transection in MS is also one of the features contributing to brain atrophy (Simon, 2005). Atrophy has come to be seen as an important aspect of MS, occurring even early in the disease, measurable over intervals as short as 1 year, and increasingly likely to be a major determinant of disease disability (Trapp et al., 1998; Simon, 2005). The related idea that MS involves neurodegeneration, quite apart from its inflammatory component, has thus been introduced in discussions of MS (Hauser and Oksenberg, 2006). But however the understanding of pathogenesis evolves, MS is increasingly understood as a progressive brain disease affecting myelin, axons, and brain volume even in those with a more benign course.
Brain atrophy in MS clearly has important neurobehavioral implications, but the specific contribution of white matter involvement to atrophy is a crucial question. Atrophy implies diffuse volume loss, but in attempting to define the specific cognitive profile of MS, the regional distribution of white matter lesions assumes more significance. In the study of Brownell and Hughes (1962), the subfrontal white matter was determined to bear the heaviest plaque burden in the brain. This predilection presumably reflects the fact that the frontal lobe is the brain’s largest, and because demyelinative lesions occur randomly in the cerebral white matter, the subfrontal white matter logically stands to be the most heavily targeted of the four lobes. Indeed, the similarity of many cognitive deficits in MS to those of other frontal lobe diseases (Filley, 2000) suggests that whereas MS is typically a diffuse brain disease, its neurobehavioral effects may reflect selective interference with frontal systems.
The neuroanatomic basis of cognitive impairment has been extensively investigated with neuroimaging, and substantial evidence over many years has supported the correlation of white matter disease burden with cognitive loss. More than a score of MRI studies have found that white matter lesion burden predicts cognitive impairment as measured neuropsychologically (Medaer et al., 1987; Franklin et al., 1988; Reischies et al., 1988; Rao et al., 1989b; Callanan et al., 1989; Anzola et al., 1990; Pozzilli et al., 1991; Swirsky-Sacchetti et al., 1992; Maurelli et al., 1992; Huber et al., 1992; Feinstein et al., 1992; Comi et al., 1993; Pugnetti et al., 1993; Feinstein, Ron, and Thompson, 1993; Arnett et al., 1994; Möller et al., 1994; Tsolaki et al., 1994; Patti et al., 1995; Ryan et al., 1996; Hohol et al., 1997; Sperling et al., 2001; Penny et al., 2010). In general, cognitive loss becomes more severe as plaques assume a more confluent appearance in the periventricular white matter, and as cerebral atrophy develops. These often-replicated conclusions leave little doubt that cerebral white matter lesions in MS have important neurobehavioral implications.
Correlations between neuropsychological test performance and white matter lesion burden, however, have been generally modest, suggesting that cognitive impairment involves more than these readily identifiable lesions (Filippi et al., 2010). MRI with higher magnet strengths such as 3.0 T improves the detection of MS neuropathology, but conventional MRI is still thought to be able to detect only some of the tissue damage in MS relevant to cognition. Early proposals, such as one suggesting a threshold cerebral white matter disease burden of around 30 cm2 as the cutoff area above which cognitive impairment is likely (Swirsky-Sacchetti et al., 1992), were soon abandoned after the realization from more advanced MRI techniques that even the NAWM is not invariably normal. Data making this point obvious came from MRS (Sarchielli et al., 1999), MTI (Filippi et al., 2000), and DTI (Filippi et al., 2010) studies of MS that all revealed abnormalities in NAWM correlating with clinical dysfunction. These observations were also confirmed by postmortem studies of MS detecting axonal loss in areas of white matter that appear to be normal (Evangelou et al., 2000). Thus microscopic white matter neuropathology can be found with MRS, MTI, and especially DTI that may add to the clinical assessment and treatment of cognitively impaired MS patients.
Of all the new neuroimaging techniques, DTI has attracted the most attention because of its unequaled capacity to image the details of white matter, including its individual tracts. Studies in MS have used DTI to identify areas of NAWM at risk for degeneration (Simon et al., 2006) or correlated with cognitive dysfunction (Rovaris et al., 2008; Roca et al., 2008; Dineen et al., 2009; Hecke et al., 2010). These studies all support a contribution of white matter damage to cognitive impairment that goes beyond that expected with white matter hyperintensities alone. Advanced neuroimaging of NAWM helps explain why the correlations of lesion volume with cognitive dysfunction in MS have not been more robust.
In spite of the mounting evidence that white matter neuropathology, both from grossly visible plaques and in the NAWM, contributes to cognitive impairment, in recent years the potential contribution of gray matter disease in MS has received much attention (Feinstein, 2007; Stadelmann et al., 2008). This possibility remains alive because of the presence of white matter fascicles in both the cerebral cortex and the subcortical gray matter, where the demyelinative process may also take hold. Most of the recent attention has been devoted to cortical plaques, and the notion of “cortical MS” has been much discussed (Stadelmann et al., 2008). Indeed, as discussed earlier, early neuropathological studies found that MS plaques could occur in the cortex, and were judged to be 5% of the total by Brownell and Hughes (1962). Such a modest disease burden would not be expected to exert a powerful effect on cogntion, and initial studies of cortical plaques with conventional MRI showed that they accounted for just 6% of the total lesion volume and did not correlate with neurocognitive test results (Catalaa et al., 1999). Study of the question has continued, however, as neurologists are attracted to the notion that the effects of MS on cognition may originate in gray matter lesions (Chapter 1).
The contribution of cortical lesions to neurobehavioral dysfunction is uncertain. Both cortical plaques and brain atrophy have been shown to be independent predictors of cognitive dysfunction (Langdon, 2011), but the visualization of cortical plaques with standard MRI field strengths is fraught with difficulty (Filippi et al., 2010). To address this problem, studies with 3.0 and 7.0 T MRI scanners are underway. As this work proceeds, it is worth keeping in mind that not all MS patients have cortical plaques, many of the plaques also involve some adjacent white matter, and gray matter inflammation is generally less extensive than in white matter (Stadelmann et al., 2008).
From a clinical perspective, it is likely that demyelination damages a higher volume of white matter in large fiber tracts than in the cortex, and the close resemblance of cognitive dysfunction in MS to that of other white matter disorders (Filley, 2012) suggests that tract demyelination is likely to be the main determinant of cognitive dysfunction, at least early in the disease. An important variable in this discussion may be the timing of neuropathology, as cortical lesions may postdate white matter lesions in the typical course of MS (Kutzelnigg et al., 2005; DeLuca et al., 2015). The neuropsychological deficits of MS patients have primarily been found in patients in early stages of the disease, and the fact that deficits resemble those of other white matter disorders implies that tract demyelination is the major determining neuropathology. Support for this idea comes from a study of pediatric MS showing that cortical lesions were rare in comparison with white matter plaques (Absinta et al., 2010). Moreover, biopsy studies of adults with early MS showed cortical demyelination in only 38% patients, while white matter lesions were present in 100% (Lucchinetti et al., 2011). In addition, while hippocampal demyelination causing synaptic alterations has been documented, this process was seen only after a mean of 26 years of disease (Dutta et al., 2011).
In general, MS is still considered a disease that predominantly affects white matter, especially early in the course (Simon, 2005; Hauser and Oksenberg, 2006; DeLuca et al., 2015). Although gray matter lesions clearly occur, they may be most significant later in the disease, and only at that time contribute to cognitive dysfunction by producing cortical involvement manifested by aphasia, apraxia, and agnosia (DeLuca et al., 2015). For now, MS surely merits inclusion on the list of diseases that can impair cognition by virtue of damage to the white matter. As the disease progresses, the cortical damage that seems to develop will require further study, and an informative research strategy would involve the longitudinal study of MS to determine if the dementia evolves from a profile of WMD to a cortical dementia as the disease progresses.
Many attempts to characterize the profile of cognitive deficits in MS have been made, and in an influential review Rao (1986) concluded that MS has neuropsychological features that qualify it as one of the subcortical dementias. The prominence of deficits in attention, concentration, memory, executive function, and neuropsychiatric status combined with the absence of significant language disturbance and other cortical deficits were seen as compelling this conclusion (Rao, 1986). Other authorities soon concurred with this opinion (Cummings, 1990). Subsequently, however, additional data were brought forth that invited the possibility that MS might have unique neuropsychological features that separate it not only from cortical diseases but also from subcortical gray matter diseases. To begin, the memory disturbance in MS was reported to involve a retrieval rather than an encoding deficit (Rao, Leo, and Aubin-Flaubert, 1989a; Brassington and Marsh, 1998; Feinstein, 2007). Whereas some evidence suggested a primary memory encoding deficit (DeLuca, Barbieri-Berger, and Johnson, 1994; DeLuca et al., 1998), and other data supported both encoding and retrieval dysfunction (Lafosse et al., 2013), the memory disorder of MS clearly differs from the encoding deficit of cortical dementia (Filley, 2012). Later, another feature that appeared to characterize MS was the sparing of procedural memory (Lafosse et al., 2007), known to be affected in subcortical gray matter diseases (Rao et al., 1993; Feinstein, 2007). These characteristics were postulated to differentiate MS from both the cortical dementias, which involve an encoding deficit, and the traditional subcortical dementias, in which procedural memory is affected; moreover, they make the case that MS may be a prototype for all the white matter dementias (Rao, 1996; Filley, 2012). Most recently, slowing of information processing speed has been emphasized and for many authorities has become a dominant feature of MS-related cognitive impairment (Feinstein, 2007; Chiaravalloti and DeLuca, 2008; Langdon, 2011). These distinctions remain the subject of systematic investigation, and further refinements will no doubt be established.
Inflammatory diseases
White matter can be the target of a diverse group of noninfectious inflammatory diseases (Box 6.4). These diseases share the common feature of autoimmune pathogenesis, and all produce systemic manifestations in addition to those in the nervous system. In the brain, widespread lesions are typical, usually related to vascular inflammation, and in many cases the clinical and neuropathological features of these diseases indicate primary damage to white matter. Many neurologic and neuroradiological similarities exist between inflammatory and demyelinative diseases (Theodoridou and Settas, 2006), and clinicians often encounter patients whose symptoms and signs invoke the possibility of differential diagnoses within both categories. Recent findings have also underscored neuropsychological commonalities in these two categories that focus on the role of white matter tract dysfunction as the source of cognitive impairment (Benedict et al., 2008). Many areas of uncertainty, however, complicate the interpretation of brain–behavior relationships in the inflammatory autoimmune brain diseases. For the purposes of this book, systemic lupus erythematosus (SLE) proves most illustrative as an inflammatory disease that often attacks white matter.
Systemic lupus erythematosus
SLE is the autoimmune connective tissue disease best known to affect the nervous system. The CNS is affected in up to two-thirds of SLE patients (West, 1994, 1996), and the PNS system may also be involved. The closely related antiphospholipid syndrome is often present in patients with SLE, and the two disorders cannot always be distinguished (Tincani et al., 2009). When SLE involves the brain, the term “lupus cerebritis” was commonly used until the more general descriptor “neuropsychiatric lupus” gained favor (West, 1994). Whereas this latter designation is useful, it includes the entire range of elemental and higher neurologic deficits, and is thus not capable of capturing the range of specific mental status alterations to which SLE patients are susceptible. To address this problem, more specific nomenclature was proposed to clarify the neurobehavioral features of neuropsychiatric lupus (ACR Ad Hoc Committee on Neuropsychiatric Lupus Nomenclature, 1999). A total of 19 neuropsychiatric syndromes were recognized, 12 of which involve the CNS; the most relevant for this account are acute confusional state, cognitive dysfunction, anxiety disorder, mood disorder, psychosis, and a demyelinating syndrome (ACR Ad Hoc Committee on Neuropsychiatric Lupus Nomenclature, 1999). As clinicians have long known, some of these syndromes can be induced by treatment of SLE, most notably with corticosteroids, but it is clear that neuropsychiatric syndromes can often develop when patients are taking small doses of corticosteroids, or none at all (Feinglass et al., 1976), pointing to cerebral involvement rather than iatrogenesis.
On the basis of autopsy data, the CNS neuropathology of SLE has been thought to be dominated by a vasculopathy with hyalinization of vessel walls and perivascular inflammation; a true vasculitis is uncommon (Johnson and Richardson, 1968). Multiple ischemic and hemorrhagic lesions in both gray and white matter result from this process (Brooks et al., 2010). However, because postmortem findings from SLE patients are usually obtained from those with severe disease, the neuropathology of more typical SLE is less well understood. Neuroradiological investigation has thus provided the main body of data on patients with milder SLE affecting the brain. Neuroimaging findings include cerebral atrophy, seen on both CT and MRI scans, but treatment with corticosteroids may confound this common finding because it is possible that atrophy may result from therapy (Jacobs et al., 1988). White matter changes, however, are also prominent, especially on MRI, and these small, multifocal lesions are the most common neuroradiological finding (Sibbitt, Sibbitt, and Brooks, 1999; Benedict et al., 2008; Toledano et al., 2013). These lesions can be distinguished from those of MS in that they display no periventricular predilection, and gadolinium enhancement is uncommon (Miller et al., 1987; Theodoridou and Settas, 2006).
The pathogenesis of neuropsychiatric dysfunction in SLE remains incompletely understood, but a variety of neuropathological processes appear to be involved. Both parenchymal damage from vascular occlusion and neuronal injury from antineuronal antibodies and cytokines have been invoked as causative factors (West, 1994, 1996; Kozora et al., 2008; Hanly et al., 2011). Neuropsychological testing has amply documented that cognitive deficits exist in some SLE patients, including both those with and those without features of neuropsychiatric lupus (Carbotte, Denburg, and Denburg, 1986; Kozora et al., 2008), and a pathogenetic role for white matter involvement has substantial support. Leukoencephalopathy seen on MRI can be associated with dementia in SLE (Kirk, Kertesz, and Polk, 1991), and fulminant, fatal leukoencephalopathy has been reported as the only feature of neuropsychiatric lupus (Prabhakaran et al., 2005).
Neuropsychological study of the profile of cognitive dysfunction in SLE generally indicates that, despite considerable clinical heterogeneity, deficits in attention, concentration, visuospatial skills, and cognitive speed without major language involvement are typical (Ginsburg et al., 1992; Kozora et al., 1996; Denburg, Carbotte, and Denburg, 1997; Kozora et al., 2008). These deficits can be seen in patients with and without other neurologic signs (Kozora et al., 1996). Fatigue, a common complaint in many patients with cerebral white matter disorders, has been correlated with MRI white matter lesion burden in SLE (Harboe et al., 2008). A study of SLE patients with the lupus anticoagulant described a profile of impairment consistent with subcortical dementia, with the most robust deficit emerging in information processing speed (Denburg et al., 1997). The usual sparing of language means that language-based screening measures such as the MMSE (Folstein, Folstein, and McHugh, 1975) are relatively insensitive to the cognitive impairments of SLE patients (ACR Ad Hoc Committee on Neuropsychiatric Lupus Nomenclature, 1999). Overall, the cognitive deficits of SLE have been interpreted as consistent with subcortical neuropathology (Denburg et al., 1997), but the pattern is also reminiscent of that produced by other white matter disorders (Filley, 2012). Consistent with this idea, a detailed neuropsychological review comparing SLE and MS found the cognitive dysfunction to be remarkably similar in the two diseases (Benedict et al., 2008).
Whereas the presence of scattered white matter lesions on conventional MRI is well known in SLE, a certain threshold burden of disease on MRI may be required to produce cognitive impairment (Kozora et al., 1998; Kozora et al., 2008). This concept implies that more subtle disease in NAWM may also contribute to cognitive loss in some patients. Thus recent research has turned to examining the microstructure of white matter to assess potential damage that cannot be detected by conventional MRI. MRS and DTI have both offered useful insights. Brooks and colleagues (1999) reported that MRS measures of neuronal dysfunction in NAWM strongly correlated with cognitive impairment: Reduced NAA implied axonal damage, and increased choline suggested inflammation and demyelination. A subsequent study of SLE patients with no prior neuropsychiatric involvement found that, in frontal white matter with no hyperintense lesions, elevated choline correlated with impaired processing speed, executive function, and sustained attention, while NAA was unaffected (Filley et al., 2009). These findings suggest that subtle myelinopathy is an early feature of the pathogenesis of cognitive dysfunction in SLE, occurring before neuropsychiatric events or white matter hyperintensities develop (Filley et al., 2009). DTI studies have shown similar findings, identifiying microstructural abnormalities in frontal and other white matter regions among SLE patients both with and without neuropsychiatric dysfunction (Zhang et al., 2007; Schmidt-Wilcke et al., 2014). An informative DTI study showed that decreased FA in a variety of normal-appearing tracts correlated with neuropsychological impairment; this relationship was more apparent for patients with neuropsychiatric SLE, and slowed processing speed was the deficit most strongly correlated with changes in FA (Jung et al., 2012). Thus early myelinopathy, likely involving immune and possibly other factors, may explain subtle neuropsychiatric dysfunction in SLE that is manifested by cognitive slowing, executive dysfunction, and impaired sustained attention (Kozora and Filley, 2011). To illustrate the complex neuropathology of this disease, MRS has also disclosed evidence of hippocampal involvement in early SLE (Kozora et al., 2011), and thus cortical involvement cannot be discounted. However, the opportunity to examine the cerebral white matter before overt neurobehavioral dysfunction begins or conventional MRI findings become apparent represents an important approach to the investigation of cognitive and emotional dysfunction in SLE. The early involvement of white matter in SLE has also been the principal impetus to work on the syndrome of mild cognitive dysfunction, which will be taken up in Chapter 8.
Infectious diseases
Infections of the brain cannot be characterized as strictly confined to the cortical, subcortical gray, or white matter, but a number of infectious diseases display a predilection for white matter (Vargas et al., 2009). These infections are caused by viruses (Box 6.5), and many clinical and immunopathological similarities to demyelinative diseases have been found when these infections are compared to MS and related disorders. As discussed with MS and SLE, infectious diseases of the white matter also have neurobehavioral consequences.
Human immunodeficiency virus infection
Progressive multifocal leukoencephalopathy
Subacute sclerosing panencephalitis
Progressive rubella panencephalitis
Varicella zoster encephalopathy
Cytomegalovirus encephalitis
Human immunodeficiency virus infection
Infection with the human immunodeficiency virus type 1 (HIV-1), commonly known as HIV, has been one of the most publicized medical problems of the past three decades. Since their recognition in 1981, the acquired immunodeficiency syndrome (AIDS) and other forms of HIV infection have become major public health problems worldwide. Despite rapid progress in elucidating its etiology, prevention, and treatment, HIV infection continues to be an incurable disease.
One of the more prominent manifestations of this systemic retroviral infection is brain involvement, which was recognized soon after the AIDS epidemic was identified (Price et al., 1988). The term first employed for this disorder was the AIDS dementia complex (ADC), which emphasized the various cognitive, motor, and behavioral changes that occur (Navia, Jordan, and Price, 1986), and other names have been subacute HIV encephalitis, AIDS-related dementia, and AIDS encephalopathy. As experience with AIDS expanded, a spectrum of cognitive impairment was appreciated, from mild cognitive dysfunction detected on neuropsychological testing (Wilkie et al., 1990) to dementia as the presenting syndrome of patients with a poor prognosis (Navia and Price, 1987; Brew, 1999). The current terminology of cognitive impairment, recognizing the wide range of dysfunction that can be seen (Antinori et al., 2007), uses the overarching term HIV-associated neurocognitive disorder (HAND), within which are included asymptomatic neurocognitive impairment (ANI), HIV-associated mild neurocognitive disorder (MND), and HIV-associated dementia (HAD).
In early studies, the ADC was found to affect approximately 30% of AIDS patients at some point in the disease (McArthur, Sacktor, and Selnes, 1998). With the introduction of antiretroviral therapy (ART) in the 1990s, impressive advances in treating cognitive dysfunction were gradually achieved (Manji, Jäger, and Winston, 2013), so that the prevalence of HAD has recently been cited as just 2% (Heaton et al., 2010). Optimism should be tempered, however, by the continued high prevalence of HAND, which is now slightly over 50% of HIV-infected individuals (Heaton et al., 2010). Whereas much progress in treatment has been made to reduce the burden of dementia, HIV infection often persists in the brain to produce some form of cognitive impairment (Gannon, Khan, and Kolson, 2011).
When first identified in the 1980s, the ADC was observed to manifest diffuse myelin pallor in the cerebral white matter as its most common neuropathological feature (Navia et al., 1986). Cases of HIV infection were reported in which white matter was severely and selectively involved, and in which fulminant fatal leukoencephalopathy was the only clinical manifestation (Jones et al., 1988; Silver et al., 1997). As is now recognized, HIV acts as a Trojan horse, entering the CNS from the bloodstream via infected monocytes to infect glial cells but not neurons (Anthony and Bell, 2008; Gannon et al., 2011). After reaching the brain, the virus is more concentrated in the white matter and basal ganglia than in the cortex (McArthur, Sacktor, and Selnes, 1999; Anthony and Bell, 2008). In typical cases of ADC, pallor of the white matter is accompanied by gliosis, multinucleated giant cells, microglial nodules, and increased numbers of perivascular macrophages (Sharer, 1992). Demyelination is not seen, and white matter pallor is thought to be due to breakdown of the blood–brain barrier and development of vasogenic edema (Power et al., 1993), or to an indirect effect of the host immune response to the virus (Anthony and Bell, 2008). Studies of the cortex in HIV infection have found either no cortical neuronal loss in ADC patients (Seilhean et al., 1993) or that the loss of cortical neurons has no correlation with dementia severity (Weis, Haug, and Budka, 1993; Everall et al., 1994). Cortical neuronal loss may occur late in the course of the disease, but white matter pallor and gliosis are more prominent initially (Gray et al., 1996; Anthony and Bell, 2008). In the ART era, white matter continues to attract neuropathological study (Everall, Hansen, and Masliah, 2005), and a recent autopsy study of HIV-infected brains showed decreased corpus callosum volume (Wohlschlaeger et al., 2009).
Neuroimaging also supports the prominence of white matter changes in HIV infection. Head CT shows some white matter involvement, and brain MRI can disclose patchy or diffuse hyperintensity on T2-weighted images along with with cerebral atrophy (Bencherif and Rottenberg, 1998). The subcortical and cortical gray matter is not affected as early or as significantly as the white matter on neuroimaging scans. MRS studies of patients with ADC suggested that frontal white matter is affected earliest, followed by the basal ganglia and then the frontal gray matter (Chang et al., 1999). Subsequent investigation in HIV patients has found that neuropsychological dysfunction is associated with abnormal MRS findings only within the white matter and not the deep gray matter (Mohamed et al., 2010).
DTI studies in HIV have extended these findings. Early work disclosed abnormalities in NAWM of HIV patients that correlated with disease severity as measured by viral load (Filippi et al., 2000). Later, microstructural damage within association tracts was found in HIV patients who were not demented (Pfefferbaum et al., 2009), and DTI changes in the corpus callosum were found to correlate with cognitive dysfunction (Wu et al., 2006). Moreover, NAWM abnormalities on DTI were found to be more severe in HAD than in nondemented HIV patients (Chen et al., 2009), and DTI global tractography metrics correlated with processing speed and executive function (Tate et al., 2010). Most recently, pervasive white matter dysfunction was found among patients early in the course of AIDS (Xuan et al., 2013), and in older adults with HIV, white matter changes correlated with summary scores of neuropsychological impairment (Nir et al., 2014).
The cognitive profile of HIV brain infection has been steadily clarified. The ADC was initially characterized as a subcortical dementia (Navia, Jordan, and Price, 1986; Tross et al., 1988), based on observations that patients typically had impairments in attention, concentration, memory, and personality, with often striking loss of cognitive speed and mental flexibility (Grant et al., 1987). Tests of choice reaction time showed marked impairment in AIDS patients (Perdices and Cooper, 1989), with usually normal language (McArthur, Sacktor, and Selnes, 1999) but impaired visuospatial function (Tross et al., 1988). Memory was uniformly affected, with a pattern of memory dysfunction rendering ADC a close fit with the hypothesized profile of WMD (Filley, 1998) because of a characteristic retrieval deficit in declarative memory (White et al., 1997) while procedural memory was spared (Jones and Tranel, 1991; Gonzalez et al., 2008). Psychomotor slowing, apathy, and withdrawal were also commonly found (Navia, Jordan, and Price, 1986). An overall relationship between the extent of white matter pallor and degree of cognitive impairment was observed in early studies (Navia et al., 1986; Grant et al., 1987; Price et al., 1988; Bencherif and Rottenberg, 1998). This association was most apparent in ADC with severe dementia, but milder cognitive impairment was also associated with changes in white matter (Post, Berger, and Quencer, 1991). Early and selective involvement of frontal white matter was suggested by a profile of neuropsychological deficits consistent with attentional impairment and executive dysfunction in mildly affected ADC patients (Krikorian and Wrobel, 1991). These observations portrayed a general trend for early white matter involvement producing subtle neurobehavioral features preceding more obvious dysfunction later on. Subsequent studies focused on cognitive slowing as a hallmark feature of HIV cognitive impairment, and the explanation for this problem was postulated to be white matter involvement (Woods et al., 2009).
Whereas the neuropathology of HIV infection has steadily implicated white matter (Navia et al., 1986), and, with further study after the introduction of ART, white matter pathology continues to be recognized (Langford et al., 2002; Langford et al., 2003), this infection does not spare the gray matter (Navia et al., 1986). Indeed, some have regarded changes in the gray matter of the basal ganglia, thalamus, and brain stem as equal to or more prominent than those in the cerebral white matter (Brew, 1999). In light of this complexity, it is problematic to determine precisely what proportion of the neurobehavioral syndrome of ADC can be attributed to white versus gray matter involvement. Some authors have dealt with this conundrum by describing the locus of HIV cognitive impairment with terms such as “frontodiencephalic” (Perdices and Cooper, 1990) and “frontostriatal” (Sahakian et al., 1995), implying that a combination of subcortical white and gray matter pathology may explain the dementia of HIV infection. Cortical involvement has also been proposed in HIV patients (Everall, Hansen, and Masliah, 2005), although one explanation is likely to be AD neuropathology given that HIV-infected persons survive longer with ART and acquire a higher risk for this disease (Brousseau et al., 2009). In view of the neuropathological heterogeneity of HIV infection, a need exists for careful studies correlating clinical features with neuroimaging and neuropathological data to improve the understanding of the origin of neurobehavioral dysfunction in these patients.
Despite the still uncertain clinical-pathological correlation in the HIV infection, further evidence of white matter dysfunction can be found in studies of ART and its effects. As discussed previously, a most welcome development in the 1990s was the marked reduction in the incidence of ADC that accompanied the widespread use of ART (Clifford et al., 2000; Manji, Jäger, and Winston, 2013). This benefit may in part have occurred through the prevention of initial cerebral white matter involvement. The antiretroviral drug zidovudine (AZT), for example, was an early mainstay of AIDS pharmacotherapy, and was shown to improve cognitive function in ADC (Sidtis et al., 1993) in parallel with an improvement accompanied by reduction in MRI white matter lesion burden (Tozzi et al., 1993; Hoogland and Portegies, 2014). Use of the protease inhibitors in the ADC was also associated with improvement in both cognitive function and the extent of white matter involvement on MRI (Filippi et al., 1998; Thurnher et al., 2000). Similarly, a recent case of acutely altered mental status with pontine demyelination in HIV infection was noted to improve both clinically and radiologically after treatment with ART (Tanioka et al., 2007). Whereas this area of inquiry has recently become complicated by concerns that newer forms of ART may be toxic to white matter (Gannon et al., 2011), it is nonetheless encouraging that recent DTI studies have shown that ART can improve white matter structural integrity after the initiation of therapy (Wright et al., 2012).
Available information, therefore, provides considerable support for the hypothesis that the neurobehavioral features of HAND, MND, and particularly HAD may be substantially related to cerebral white matter involvement. This conclusion does not preclude a contribution of changes in gray matter, subcortical and perhaps even cortical, to the clinical presentation. Moreover, the complexity of diffuse metabolic dysfunction in HIV infection, most likely involving a variety of neurotoxic inflammatory mediators (McArthur, Sacktor, and Selnes, 1999; Gannon et al., 2011), should also be considered. Among the factors involved in the pathogenesis of cognitive dysfunction in HIV brain infection, however, white matter dysfunction must be included.
Metabolic disorders
Several disorders in the general category of metabolic dysfunction can result in white matter disease of the brain (Box 6.6). Although considerable overlap exists between metabolic and toxic disorders (Hinchey et al., 1996), the conditions in Box 6.6 can be seen as stemming from a metabolic derangement in which clinical, neuroimaging, or neuropathological evidence of leukoencephalopathy has been observed. The pathophysiology of these diverse disorders is incompletely understood in most cases, but a disturbance of brain metabolism is present in each. Returning to a theme common in the white matter disorders, many of these disorders are reversible if the metabolic derangement is corrected before irreversible damage develops. Neurobehavioral aspects of these disorders have been gradually characterized and consistent findings have emerged, although more study is warranted in the pursuit of specific correlations between well-defined white matter lesions and neurobehavioral features.
Cobalamin (vitamin B12) deficiency
Folate deficiency
Central pontine myelinolysis
Hypoxia
Hypertensive encephalopathy
Eclampsia
High-altitude cerebral edema
Cobalamin deficiency
For our purposes, the most useful example of this category is dementia related to cobalamin (vitamin B12) deficiency. Cobalamin, routinely measured in the evaluation of neurologic patients, is important in the maintenance of normal myelin, and its deficiency results in the well-known spinal cord syndrome known as subacute combined degeneration (Stabler, 2013). Far less well recognized is that cobalamin deficiency may also cause perivascular degeneration of myelinated fibers in the cerebrum that is identical to the spinal cord pathology in subacute combined degeneration, and these brain lesions have been postulated to account for dementia (Adams and Kubik, 1944). Cobalamin deficiency has been noted in up to 40% of older individuals, and in those who are deficient, as many as 50% may have symptoms referable to cerebral involvement (Goebels and Soyka, 2000). Classically associated with pernicious anemia, it has become clear that neurologically significant cobalamin deficiency can occur in patients with neither anemia nor macrocytosis (Lindenbaum et al., 1988). Thus the routine screening of serum vitamin B12 levels in older persons has been recommended (Pennypacker et al., 1992). Some uncertainty exists, however, about the level of cobalamin that indicates significant tissue depletion. Levels below 100 pg/ml are widely thought to produce neurologic mainfestations, and those above 300 pg/ml are regarded by most authorities as normal. B12 levels that fall within the 100–300 pg/ml range are intermediate, and many recommend the measurement of serum homocysteine and methlymalonic acid in these patients; if one or both of these metabolites are elevated above the normal range, clinically significant cobalamin deficiency can be assumed (Pennypacker et al., 1992; Stabler, 2013). In patients who are found to be cobalamin deficient, peripheral neuropathy is the most common neurologic syndrome, affecting 70% of those with neurologic complaints (Healton et al., 1991). Clinicians are also well aware of the myelopathy of subacute combined degeneration, but the specific cerebral manifestations of vitamin B12 deficiency are not as familiar.
The neurobehavioral manifestations of cobalamin deficiency are generally considered to be protean. Neuropsychiatric dysfunction has often been described (Shorvon et al., 1980), and psychosis appears to be especially common (Hutto, 1997). A study of community-dwelling older women found that metabolically significant cobalamin deficiency increased the risk of severe depression twofold (Penninx et al., 2000). Cognitive loss and dementia have both been documented (Meadows, Kaplan, and Bromfield, 1994; Larner et al., 1999), and the profile of deficits in these syndromes, which may often include cognitive slowing and confusion along with depression, has been interpreted as consistent with subcortical dementia (Tenuisse et al., 1996; Larner et al., 1999). Consistent with this conclusion, Osimani and colleagues (2005) found that B12 deficiency produced a reversible dementia characterized by early psychosis and impaired concentration, visuospatial performance, and executive function without language disturbance.
Neuropathological observations of the brain in cobalamin deficiency have long been available (Woltman, 1918; Adams and Kubik 1944), but many clinicians may not associate this disorder with cerebral white matter involvement. The brain lesions usually develop later in the course than spinal cord lesions, and are characterized by perivascular degeneration of myelinated fibers with sparing of cortical and subcortical gray matter (Adams and Kubik, 1944). Laboratory experiments have shown that monkeys deprived of cobalamin have white matter changes in the cerebrum identical to those seen in B12-deficient humans (Agamanolis et al., 1976). The underlying pathophysiology of cobalamin deficiency is thought to involve a disturbance of fatty acid synthesis leading to abnormal myelination (Shevell and Rosenblatt, 1992; Smith and Refsum, 2009). More recently, vascular mechanisms of injury have also been implicated (Tangney et al., 2011).
In the mid-twentieth century, the cerebral white matter lesions seen neuropathologically were proposed to be responsible for the mental status changes in patients with cobalamin deficiency (Adams and Kubik, 1944), and MRI studies have consistently supported this claim. Low cobalamin levels are associated with the severity of MRI white matter lesions (de Lau et al., 2009; Tangney et al., 2011), and several case studies have noted clinical and neuroradiological improvement of leukoencephalopathy occurring in parallel with cobalamin replacement (Chatterjee et al., 1996; Stojsavljević et al., 1997; Vry et al., 2005). A 2-year follow-up study showed that low B12 levels were associated with progression of periventricular white matter hyperintensities in lacunar stroke patients (van Overbeek, Staals, and van Oostenbrugge, 2013). A recent literature review concluded that cobalamin deficiency is associated with cognitive impairment, white matter damage, and brain atrophy (Smith and Refsum, 2009).
A variety of pathogenic disturbances occur in the brain and may contribute to the neurobehavioral aspects of this disorder (Penninx et al., 2000). The biochemistry of cobalamin metabolism is complex, and many factors may contribute to the development of dementia. One that has attracted much recent attention is the effect of hyperhomocysteinemia, which is strongly associated with clinically significant cobalamin deficiency (Lindenbaum et al., 1988). Homocysteine has been recognized as an independent risk factor for cerebrovascular disease (Fassbender et al., 1999), and white matter lesions related to elevated homocysteine have been documented (Wright et al., 2005). These considerations further suggest that vascular as well as nonvascular mechanisms are involved in the pathogenesis of cobalamin deficiency (Tangney et al., 2011).
Cobalamin replacement has been observed to benefit neurobehavioral syndromes in some individuals with vitamin B12 deficiency. The extent of true reversibility has been debated, as some authorities describe very few responders (Clarfield, 1988), whereas others report a substantial number (Lindenbaum et al., 1988). It is also not entirely clear how significant is the recovery that actually takes place in patients who are reportedly improved. Nevertheless, well-documented examples of meaningful cognitive and neuroimaging recovery with cobalamin treatment (Meadows, Kaplan, and Bromfield, 1994; Chatterjee et al., 1996; Stojsavljević et al., 1997; Vry et al., 2005) suggest that reversible dementia results from white matter involvement in some patients with cobalamin deficiency. Further studies using clinical and neuroimaging measures with larger numbers of subjects will be necessary to expand on these findings. In this regard, encouraging data were recently presented from a community-based European study of older people demonstrating that regular intake of B12 reduced the conversion of mild cognitive impairment to dementia, and produced lower grades of MRI white matter hyperintensity (Blasko et al., 2012). The evidence to date thus favors a salutary effect of cobalamin on white matter, and maintenance of at least adequate dietary cobalamin intake has been recommended, especially for older individuals (Smith and Refsum, 2009).
Hydrocephalus
Hydrocephalus refers to the accumulation of excessive water in the cranium (Box 6.7). This added volume is associated with ventricular enlargement, particularly involving the lateral ventricles, and the effects on adjacent brain structures may be relevant for neurobehavioral function. Normally the total volume of cerebrospinal fluid (CSF) within the neuraxis is about 140 cm3, of which about 25 cm3 are found in the four ventricles (Fishman, 1992). Higher ventricular volumes of CSF can develop either because additional CSF occupies intracranial space and compresses brain tissue or because the CSF replaces the parenchyma lost through brain atrophy (hydrocephalus ex vacuo). The cerebral white matter is implicated in both of these situations, because either an excessive volume of CSF may injure the periventricular regions, or changes in white matter contribute to cerebral atrophy.
Hydrocephalus can be symptomatic at any age, and its most prominent neuropathological effects are exerted on cerebral white matter (Di Rocco et al., 1977; Akai et al., 1987; Del Bigio, 1993; Del Bigio et al., 1994; Leinonen et al., 2012). Cortical damage is uncommon, occurring only late in the disease course, and involvement of the deep gray matter is also less marked than white matter injury, indicating that the cognitive effects of hydrocephalus are mainly related to tract damage, at least when diagnostic and treatment issues are most crucial. Hydrocephalus occurring acutely in the context of mass lesions, trauma, infarction, and the like will not be reviewed here, because of methodological difficulties in studying the impact of specific white matter neuropathology in these settings. Much useful information, however, has been gained from study of hydrocephalus that arises chronically. In those with chronic sequelae of hydrocephalus, the medical and economic impact can be substantial, and the problem of hydrocephalus in older people represents an especially challenging diagnostic and therapeutic problem in behavioral neurology (Del Bigio, 1998; Shprecher, Schwalb, and Kurlan, 2008; Wilson and Williams, 2010).
Normal pressure hydrocephalus
Few conditions in clinical neurology produce such diagnostic, pathophysiological, and therapeutic perplexity as normal pressure hydrocephalus (NPH). Since the first description of this disease in the 1960s (Adams et al., 1965), NPH has alternately been heralded as an often-overlooked reversible dementia and a questionable entity that may not even exist. While neurologists generally agree that NPH is a diagnosable disease, and the opportunity to effectively treat a dementia syndrome is of course appealing, the vagaries of diagnosis and the risks of surgical treatment are not insubstantial. When patients present with symptoms and signs suggesting NPH, the neurologist is thus challenged to decide upon which individuals actually have the disease and, of these, which ones are likely to benefit from surgical treatment. Such patients are not infrequently seen in neurologic practice, and it has been estimated that 1% to 5% of older people with dementia may have NPH (Wilson and Williams, 2010). The continuing clinical conundrum of this disease will be discussed here in light of evidence that it can be interpreted as a disorder of cerebral white matter.
NPH classically presents with the clinical triad of dementia, gait disorder, and urinary incontinence (Adams et al., 1965). All of these problems were related to frontal lobe damage in the original description of the disease, and cognitive slowing and apathy were called out as prominent features of the dementia (Adams et al., 1965). More recent investigations have led to a consensus that the neuroanatomic alterations of NPH involve the periventricular white matter and disrupt frontal-subcortical circuits (Del Bigio, 2010; Wilson and Williams, 2010). Some cases are thought to follow meningitis, traumatic brain injury, or subarachnoid hemorrhage, but many remain idiopathic despite thorough investigation (Graff-Radford, 1999). CT scans show enlarged ventricles and relatively normal cortical gyri without sulcal enlargement, findings that offer some help but are often difficult to distinguish from other neuropathological states and normal aging.
MRI may reveal additional high signal lesions in the periventricular white matter, and it has been proposed that transpendymal CSF flow is a clue to a good outcome with shunting (Jack et al., 1987). Other investigations have not been consistent in supporting this claim (Graff-Radford, 1999). At present, it is thought that no neuroimaging finding can be used to predict outcome after shunt surgery (Wilson and Williams, 2010). MTI studies have shown a low MTR in the NAWM of patients with NPH (Hähnel et al., 2000), and recent DTI studies have demonstrated improvement in corona radiata FA after shunt surgery or ventriculostomy (Assaf et al., 2006). Data such as these may assist in demonstrating white matter damage in NPH, and its restitution after treatment.
The diagnosis is currently made by identifying the clinical triad, finding a consistent neuroimaging pattern, and ruling out other etiologic possibilities; many clinicians also perform a high-volume CSF “tap test” because a temporary improvement in gait and sometimes cognition may help predict a good surgical outcome (Fisher, 1978). Among patients accurately diagnosed with NPH, 46% to 64% may have a favorable response to shunt surgery, and the gait disorder usually responds better than dementia and urinary dysfunction (Wilson and Williams, 2010). The favorable response to shunt surgery suggests that damaged white matter in NPH may exhibit some degree of plasticity, especially if the disease is treated at an early stage.
The pathophysiology of NPH continues to be a puzzle. Adams and colleagues (1965) offered the first and still most familiar theory, proposing that obstruction of CSF outflow causes ventricular enlargement without cortical atrophy; the assumption here is that the outflow obstruction is at the level of the arachnoid villi. The CSF pressure remains normal because of Pascal’s law (Force = Pressure × Area), which predicts that with an increased ventricular wall area, the force applied to the brain parenchyma can be high while pressure is still normal (Adams et al., 1965). This explanation, however, has been questioned by some. Symon and colleagues (Symon, Dorsch, and Stephens, 1972) found episodes of raised intracranial pressure throughout the night in patients with NPH, and suggested that a better term for the condition might be “episodically raised pressure hydrocephalus.” Geschwind (1968) questioned the theory of Adams and colleagues on physical grounds, pointing out that the properties of the ventricular wall structure pertain to the pathogenesis of NPH. These ideas helped lead to an alternative hypothesis, which is that ischemia and infarction in the cerebral white matter may lead to ventriculomegaly, and the reduced tensile strength of the white matter could cause further ventricular enlargement under the stress of intraventricular pulse pressure (Earnest et al., 1974). In support of this notion is the finding of reduced cerebral blood flow in the white matter of NPH patients, most notably in the periventricular regions (Momjian et al., 2004). This theory thus suggests a possible overlap of NPH with BD (Wilson and Williams, 2010).
Most authorities, however, still favor the existence of NPH as a discrete entity, recognizing that it may coexist with other neuropathologies. The major comorbid neuropathology of NPH within the white matter is BD, but the two diseases can be distinguished at the microstructural level. CSF sulfatide is markedly increased in BD, consistent with ischemic demyelination (Tullberg et al., 2000), and white matter axons are damaged in BD but relatively intact in NPH, helping explain why BD has a worse prognosis (Tullberg et al., 2009). Moreover, whereas MRI white matter lesions in NPH may be very similar to those of BD (Tullberg et al., 2002), NPH patients without comorbid vascular white matter disease have a better outcome after shunt surgery (Boon et al., 1999; Tullberg et al., 2000). Clinically, the beneficial response to surgery seen in many NPH patients seems to justify the conclusion that the disease may exist in isolation, but that white matter ischemia and infarction can often be present in addition and limit its reversibility.
Regardless of the pathogenetic mechanism of NPH, neuropathological findings are generally consistent with primary involvement of white matter. Details of the neuropathology have been clarified over many years of investigation, and the cerebral white matter bears the brunt of the damage (Di Rocco et al., 1977; Akai et al., 1987; Del Bigio, 1993; Del Bigio et al., 1994; Del Bigio, 1998; Del Bigio, Wilson, and Enno, 2003; Del Bigio and Enno, 2008; Del Bigio, 2010; Leinonen et al., 2012). The corpus callosum is affected, and its effacement can be seen well on sagittal MRI images (Wilson and Williams, 2010). In the periventricular white matter, evidence exists for direct mechanical compression (Di Rocco et al., 1977; Del Bigio, 2010) as well as ischemic demyelination and infarction (Akai et al., 1987). Gray matter, meanwhile, both of the subcortical nuclear structures and of the cerebral cortex, is less affected, although with severe and prolonged hydrocephalus there may be some neuronal damage in these regions (Del Bigio, 1993; Del Bigio, Wilson, and Enno, 2003; Del Bigio, 2010; Leinonen et al., 2012). As is true of other white matter disorders, however, unrelated neuropathology in the cortex can be found in patients with NPH. One group addressed this issue, and found that 10 of 38 patients thought clinically to have NPH actually had biopsy-proven AD (Bech et al., 1997). BD may also be present in the white matter and compromise recovery after shunting procedures (Wilson and Williams 2010).
The neurobehavioral profile of NPH has been studied in increasing detail. Comprehensive literature reviews have concluded that NPH exhibits neuropsychological features typical of subcortical dementia (Derix, 1994; Devito et al., 2005). Prominent frontal manifestations are seen, including cognitive slowing, inattention, and perseveration, and while memory loss occurs, aphasia, apraxia, and agnosia are not observed (Derix, 1994; Devito et al., 2005). Cognitive slowing, executive dysfunction, impaired attention, poor memory, apathy, and depression have all been noted in NPH patients (Gustafson and Hagberg, 1978; Ogino et al., 2006; Chaudhry et al., 2007; Hellström et al., 2008). Gallassi and colleagues (1991) compared NPH with BD patients and found these groups to have similar cognitive impairment in comparison to control subjects. Later, Iddon and colleagues (1999) demonstrated selective executive function deficits in patients with NPH that were not present in AD patients with comparable dementia severity. A recent DTI study of NPH found that scores on the Frontal Assessment Battery – a measure sensitive to executive dysfunction (Dubois et al., 2000) – correlated with microstructural abnormalities in the frontal and parietal white matter (Kanno et al., 2011). Summarizing the existing literature, Wilson and Williams (2010) concluded that frontal and subcortical systems are primarily involved in this disease. Thus, in light of demonstrable white matter involvement and the similarity of cognitive deficits with those produced by other white matter disorders, NPH merits classification as a white matter disorder capable of causing dementia (Filley, 2012).
The treatment of NPH is surgical, and involves placement of a diversionary shunt in the brain to reduce ventricular volume. These shunts are rubber tubes with one-way valves that can be positioned to provide ventriculoperitoneal, ventriculoatrial, or lumboperitoneal CSF drainage. The critical clinical issue revolves around the decision as to which patients are most likely to benefit from a shunting procedure. This determination is not trivial, particularly in older patients, because of the high incidence of shunt surgery complications such as anesthesia reactions, intracranial hemorrhage, and infection (Vanneste et al., 1992). Although clinicians have individual preferences, it is reasonable to refer for surgery only those patients who have the full clinical triad, neuroimaging studies consistent with NPH, a positive tap test, no evidence of other cerebral diseases, and no prohibitive surgical risk. Graff-Radford (1999) has suggested that additional favorable prognostic signs are the presence of dementia for less than 2 years, a known cause of NPH, and the gait abnormality preceding the dementia.
Gait disorder is viewed by most authorities as the feature of NPH most responsive to surgical treatment (Wilson and Williams, 2010; Klassen and Ahlskog, 2011). A recent controlled trial of shunted NPH patients, however, demonstrated improvements in cognitive speed and sustained attention as well as gait (Katzen et al., 2011). The improvement in cognition may result from restoration of white matter integrity, as both MRI (Tullberg et al., 2002) and DTI (Akiguchi et al., 2008) studies have suggested. Another study found that neurologic improvement after the tap test occurred in association with higher white matter integrity as assessed by DTI (Demura et al., 2012), indicating that even temporary CSF drainage may improve the structure of white matter. Last, an intriguing preliminary study of a medical treatment for NPH found that acetazolamide reduced periventricular white matter hyperintensity burden and even improved gait in some subjects (Alperin et al., 2014). These observations add further support to the contention that dementia in NPH is a result of cerebral white matter damage.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


