Chapter 55 Anoxic-Ischemic Encephalopathy
When the heart stops and cerebral blood flow is interrupted, patients lose consciousness and may remain comatose after resumption of circulation. Such a global injury to the brain is understandably profound, and more than 70% of patients die or remain comatose 24 hours after cardiopulmonary resuscitation (CPR) (Rogove et al., 1995; Zandbergen et al., 2006). Anoxic-ischemic injury—albeit less well defined and less clearly understood—may also occur in patients with respiratory arrest or severe hypoxemia (e.g., asphyxia) and in shock. Success of intervention in these conditions may be predicated on early correction of hypoxemia and hypovolemia. The time interval until correction may be less important than the initial severity of the abnormality.
Approximately 100,000 patients a year in the United States are admitted to intensive care units with anoxic-ischemic brain injury after CPR (Peberdy et al., 2003). Although the pathophysiology of brain injury caused by cardiac arrest is well understood, less is known about neuroprotection. There is, however, some hope that induced moderate hypothermia could not only affect survival but also improve neurological outcome if patients are immediately subjected to cooling (Broccard, 2006). This chapter critically evaluates the current knowledge of anoxic-ischemic brain injury after cardiac and respiratory arrest. Studies have reported tools for predicting outcomes, and guidelines for prediction of poor outcome have been developed by the American Academy of Neurology (Wijdicks et al., 2006). The accuracy of these predictors after the use of therapeutic hypothermia is a subject of ongoing investigations. Delayed postanoxic deterioration caused by carbon monoxide intoxication is discussed in Chapter 58.
Pathophysiological Concepts
One of the more vital questions for scientists and clinicians is whether there is a specific period during which these patients can be effectively treated. Is the damage to the brain permanent and present at ictus, or are there processes at work that could potentially be influenced and modulated? Several clinical facts are important. First, with cardiac arrest, whether due to asystole or ventricular fibrillation, there is no measurable flow to the brain. Moreover, even with standard CPR techniques, only one-third of the pre-arrest cerebral blood flow can be attained (Maramattom et al., 2005). In addition, the shockable rhythms (ventricular tachycardia and ventricular fibrillation) have a better outcome than “nonshockable” rhythms such as asystole and bradyarrhythmias, reflected by restoration of adequate cerebral blood flow when ejection fraction of the ventricle improves (Callans, 2004). Secondly, there might be a critical time period after which CPR may fail to restore neuronal function. This time interval is poorly defined, but we know that the neuronal oxygen stores are depleted within 20 seconds of cardiac arrest, and cerebral necrosis occurs as a result of ischemia. There is some uncertainty about whether hypoxemia alone could produce necrosis and, although it can cause damage (preferentially in the striatum), necrosis is rarely seen even in patients with arterial Po2 values less than 20 mm Hg.
After the brain has been injured by anoxic-ischemic insult, several biochemical mechanisms may become operative (Fig. 55.1). Selective neuronal vulnerability to this type of injury involves areas in the hippocampus, the CA-1 sector, the thalami, the neocortex, and the Purkinje cells (Fig. 55.2). Necrosis of the cortex involves layers three, four, and five and is pathologically known as laminar necrosis. The vulnerability of these areas may be explained by the presence of receptors for excitatory neurotransmitters or the high metabolic demands of these neurons. An important question is whether necrosis or apoptosis occurs. The cell death cascade that involves several modulatory and degradation signals has been documented in global cerebral ischemia, but whether manipulation of these processes is effective remains unclear (Ogawa, 2007). A caspase inhibitor did not affect neurological outcome after 6 minutes of cardiopulmonary arrest in rats (Teschendorf et al., 2001).
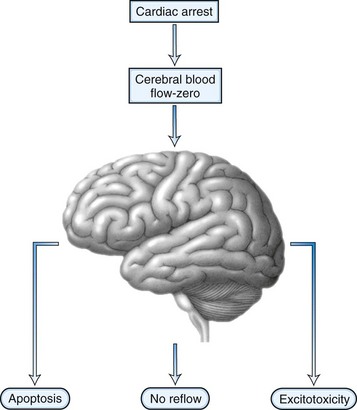
Another mechanism of damage is excitatory brain injury. Glutamate efflux due to ischemic injury increases intracellular calcium concentration, which results in neuronal injury. The excess release of calcium leads to other processes that include activation of catabolic enzymes and endonucleases. Glutamate excitotoxicity has remained the major hypothesis to explain the neuronal injury and was made more probable after the documentation of neuroprotection with N-methyl-d-aspartate (NMDA) or α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor antagonists. Research interest has pointed toward a phenomenon called no reflow. This concept is based on the premise that after resumption of circulation, there are major microcirculatory reperfusion deficits. Coagulation may occur within these reperfusion zones, with intravascular fibrin formation and microthrombosis; this concept is currently an incentive for a reperfusion trial using recombinant tissue-type plasminogen activator (tPA). Also, the use of hypertonic hyperoncotic solutions improved these perfusion deficits. Despite our understanding of the pathophysiology of anoxic-ischemic injury based on careful animal experiments, the clinical reality is discouraging. Clinical trials using barbiturates or calcium channel antagonists have been unsuccessful (Maramattom and Wijdicks, 2005). Induced hypothermia, which inhibits apoptosis and reduces free radical formation and excitatory neurotransmitters, might be the only current beneficial intervention (Bernard et al., 2002; Group THACAS, 2002). Patients who are comatose after CPR unfortunately often have a devastating outcome. Improvement of outcome might come from very early intervention and administration of neuroprotective agents at the onset of resuscitation, rather than when a patient enters the hospital.
Clinical Examination
Early awakening after CPR, clinical signs that indicate localization of the pain stimuli, or following simple commands are indicators of a good outcome. However, the current literature provides no criteria on which a good outcome can be based. Most studies have specifically concentrated on the examination of the patient, assuming a poor outcome. An estimated 25% of patients remain comatose in the first 24 hours. The mortality rate in patients who have been resuscitated following cardiopulmonary arrest approaches 80% to 90% when they have not awakened within the first 24 hours (Zandbergen et al., 2006).
Clinical neurological examination follows a standard procedure, with examination of the brainstem reflexes, motor response to pain, specific attention to myoclonus, and spontaneous or elicited eye movement abnormalities. Because the brainstem is far more resilient to anoxic-ischemic injury than the cortex, brainstem reflexes, including the pupillary reflex to light, are often normal. Absent pupil responses can be caused by a high dose of intravenous atropine used during resuscitation, although a pupil response can often still be found when examined under the magnifying glass. Fixed, dilated pupils presenting 6 hours after resuscitation are a sign of poor prognosis, but this is rarely present in isolation and is usually an indication that the brainstem has also been involved in the anoxic-ischemic injury. Corneal reflexes have been absent in about a third of patients, but they often reappear soon. Far more important is the presence of eye movement abnormalities (Wijdicks, 2002). Sustained upward gaze is indicative of a significant global bihemispheric injury that may include the thalamus. In some patients, downward gaze can be elicited using rapid head shaking or attempting to elicit a vestibular ocular response (Johkura et al., 2004). Other eye abnormalities, including ping-pong gaze or periodic lateral gaze deviations, have not been specifically examined for their prognostic value (Diesing and Wijdicks, 2004). Continuous blinking is often a common finding in comatose patients, although its anatomical substrate is unknown. An important clinical sign is myoclonus status epilepticus, defined as continuous jerking that involves the facial muscles, limbs, and abdominal muscles (Thomke et al., 2005; Young et al., 2005). These jerks can often be elicited by touch or hand clap and may also involve the diaphragm, which complicates ventilation. Myoclonus status epilepticus is an agonal phenomenon indicating a very poor prognosis. A high percentage of these patients have a burst suppression pattern on electroencephalogram (EEG) and computed tomography (CT), indicating either cerebral infarction or cerebral edema. Myoclonus status epilepticus must be distinguished from myoclonus due to intoxication or hepatic encephalopathy and from generalized tonic-clonic seizures. Convulsive status epilepticus is uncommon, as is nonconvulsive status. The motor response to pain should be classified and described as absence to pain, extensor response, pathological flexion response, or no motor response.
The outcomes for patients in coma range from death, including brain death to persistent vegetative state (see Chapters 5 and 32A), to awakening with disabilities ranging from the minimally conscious state (see Chapter 5) to complete recovery (see Chapter 48).
Awakening from coma can be protracted and prolonged, although the vast majority of patients awaken within the first 48 hours. In our series of patients, 94 of 101 patients with postanoxic ischemic injury awoke within 3 days after cardiac arrest and induced hypothermia did not seem to directly influence this (Fugate et al., 2011). However, awakening can occur even 3 months after onset, although rarely without a severe deficit such as an amnesic syndrome or other neurological findings (Table 55.1).
Table 55.1 Clinical Syndromes after Postanoxic-Ischemic Encephalopathy
| Clinical Syndrome | Mechanism | Outcome |
|---|---|---|
| “Man-in-the-barrel” syndrome | Bilateral watershed infarcts | Uncertain, may improve substantially |
| Parkinsonism | Infarcts in the striatum | Improvement possible |
| Action myoclonus | Cerebellar infarcts | In awake patients, could improve with medication |
The neurological examination can be confounded by an additional systemic injury associated with CPR. Several patients may have an associated acute renal failure or liver injury. In addition, medications may have been administered to counter pain or to facilitate mechanical ventilation. Often patients have been treated with fentanyl and lorazepam, both of which have long elimination half-lives (Table 55.2). The use of therapeutic hypothermia may further contribute, as hepatic metabolism and renal clearance are decreased, which may cause an enhanced and prolonged effect of medications (Polderman, 2009).
| Agent | Elimination Half-Life (Hours) |
|---|---|
| Morphine | 1.5-4 |
| Fentanyl | 2-5 |
| Alfentanil | 1.5-3.5 |
| Midazolam | 1-4 |
| Lorazepam | 10-20 |
| Propofol | 2 |








