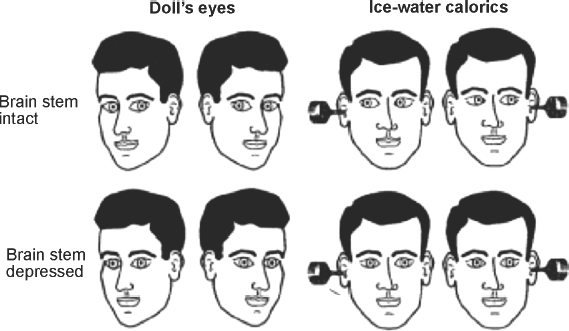
1 Assessment of Acute Loss of Consciousness Michael P. Merchut and José Biller Coma is the extreme state of unconsciousness, with an apparently “sleeping” patient unresponsive to even painful stimuli. The relevant pathophysiology was perhaps most capably reviewed by Plum and Posner1 in their landmark monograph Stupor and Coma. The clinical approach to patients in coma is emphasized in this chapter. Wakefulness or alertness is critically dependent upon the “ascending reticular activating system (ARAS),” a functional component of the complex neuronal network within the reticular formation of the upper brainstem. The ARAS extends from the tegmental mid-pons to midbrain to thalamic intralaminar nuclei and basal forebrain, from which there are widespread cortical projections, especially to the frontal lobes and limbic system.2 Large structural lesions directly interrupting this pathway (e.g., pontine infarction from basilar artery occlusion) would cause unconsciousness, whereas a unilateral, hemispheral lesion (e.g., a frontal lobe embolic infarction) would not do so, unless it indirectly impaired the ARAS by means of pressure effect. Typically, the latter occurs in the setting of severe, perilesional edema, creating a midline shift which compresses the thalamic intralaminar nuclei of the diencephalon. In the absence of midline shift or pressure on the diencephalon, coma could be produced by extensive, bilateral lesions (e.g., severe head trauma) destroying widespread cortex or disrupting most of the thalamocortical or cortico-cortical projections.3 Metabolic processes producing coma generally do so by impairing bilateral or diffuse areas of cerebral cortex, which are far more sensitive to hypoxia, hypoglycemia, and drug effects than the brainstem. Some patients appear awake, but are not attentive or responsive. Such akinetic mutism4,5 may be due to bilateral lesions in the thalamus or medial frontal lobes, particularly the anterior cingulate gyrus. After the ARAS terminates in the intralaminar thalamic nuclei, many of the thalamocortical projections terminate in the anterior cingulate, where stimulus discrimination occurs, focusing attention and responses to select stimuli, although ignoring others. The chronic or persistent vegetative state is another setting where the patient seems intermittently awake, has reflexive activity, but lacks higher cortical responsiveness to stimuli, including attempts to communicate. Coma may occur after resuscitation for cardiopulmonary arrest, and in the absence of lethal medical complications, the patient may survive quite some time with a static, anoxic encephalopathy, evolving days later into a chronic vegetative state. On the other hand, life-threatening, progressive disorders (e.g., cerebral metastases, hemorrhage) also produce coma and may soon lead to a fatal outcome if not recognized and treated early. The cause of coma in a patient must thus be diagnosed rapidly and any potential treatment begun as soon as possible. Because the clinical bedside neurological examination of the comatose patient is limited to an assessment of brainstem reflexes, the patient’s history, when available, becomes a valuable piece of information. The rapidity and manner in which the patient became comatose, as well as past medical history, current medications or procedures, and recent symptoms or illness, may all provide important clues as to the cause of coma. Observers, family, or friends noting the sudden onset of coma in an otherwise stable patient offers a strong suggestion of intracranial hemorrhage, extensive brainstem infarction or multiple cerebral embolic infarcts, or cerebral hypoperfusion from cardiac arrhythmia or heart block. Many patients with arrhythmogenic syncope awaken readily, without the postictal confusion and somnolence following a seizure. “Sudden death” occurs after refractory ventricular arrhythmias or asystole. Persistent coma after transient arrhythmia, heart block, or successful cardiopulmonary resuscitation implies some anoxic insult to the brain, with variable recovery over time. The report of an excruciating headache just prior to loss of consciousness points toward aneurysmal rupture with subarachnoid hemorrhage. Metabolic disorders producing coma usually are preceded by a period of confusion or delirium, if that history is obtainable, although drug overdosage, whether accidental or intentional, or medicinal side effects of somnolence or sedation, may also occur abruptly. New medications or dose adjustments should be scrutinized, and any “med alert” bracelets checked for a history of epilepsy, diabetes, or warfarin use. Serious head or cervical spine trauma are suggested by where the patient is found(e.g., the foot of the stairs) or by signs of external injury, if eyewitnesses are not present. A preceding febrile illness makes meningoencephalitis or brain abscess more likely in a comatose patient, although signs of infection may be lacking in the immunocompromised. While any historical clues are being obtained, other medical personnel must immediately ensure the adequacy of the “ABC’s” (airway, breathing, circulation) in the comatose patient. The vital signs themselves may suggest the cause of coma. Hypoventilation may signal direct involvement of vital medullary cardiorespiratory centers by an extensive brainstem hemorrhage or infarction, or indirectly by means of tonsillar herniation from cerebral or cerebellar edema. In the setting of head trauma, apnea may be due to an unsuspected, severe, high cervical spinal cord injury. Acute neuromuscular disorders leading to global paralysis (e.g., myasthenic crisis, severe Guillain-Barré syndrome) may also cause respiratory weakness, although those patients typically remain conscious despite having the “locked-in syndrome.” Increased blood pressure is a frequent reactive phenomenon in the setting of acute cerebral hemorrhage or infarction without impaired consciousness, but extremely elevated blood pressure may actually reflect the primary cause of coma: hypertensive encephalopathy, or a thalamic or basal ganglia hemorrhage from uncontrolled essential hypertension or sympathomimetic drug use (e.g., cocaine). In the setting of rising brain edema with incipient tonsillar herniation, an abrupt rise in blood pressure may be accompanied by bradycardia and decreased respirations: Cushing’s reflex. Initially hypotensive comatose patients either have severe hypovolemia, septicemia, cardiogenic shock, or drug-induced cardiac depression. Febrile comatose patients most often have systemic or central nervous system (CNS) infections. Other causes of fever, as suggested by the clinical scenario present, include malignant hyperthermia from anesthesia, neuroleptic malignant syndrome, serotonin syndrome, heatstroke, and anticholinergic overdose. Patients found outdoors in winter, however, may be comatose from hypothermia, which requires urgent rewarming. If the body core temperature drops further below 35°C, even brainstem reflexes may be lost and the patient appears to be brain dead. Certain features of the general physical examination may also help with the etiology of coma. Cutaneous periorbital (“raccoon eyes”) and mastoid (“Battle’s sign”) hemorrhages reflect skull fractures, as do CSF otorrhea and rhinorrhea, incriminating head trauma as the prime cause of coma in someone “found down on the ground.”6 A jaundiced patient with gross ascites may be in hepatic coma. Diffuse petechiae and ecchymoses may point toward a systemic coagulopathy and the likelihood of an intracranial hemorrhage, whereas “palpable purpura” is a sign of meningococcemia and coexistent meningitis. Nuchal rigidity may develop from meningeal infection or subarachnoid bleeding, but may be undetectable when the patient is fully comatose.7 If there is any question of trauma, however, the neck and head should not be rotated or flexed until a cervical spine fracture or instability is radiographically excluded. A preretinal or subhyaloid hemorrhage on funduscopic examination is associated with a subarachnoid hemorrhage.7 The discovery of papilledema confirms elevated intracranial pressure of diverse etiology, but takes at least 2 to 4 hours to develop after an acute cerebral or subarachnoid hemorrhage.8 Subungual (splinter), palmar, plantar, or retinal (Roth spot) hemorrhages suggest infective endocarditis as the source of cerebral emboli or abscesses in a comatose patient with a heart murmur. Although the neurological examination of comatose patients is limited, it correlates with the level of dysfunction in the CNS, and changes serially with any rostral to caudal progression of edema, hemorrhage, or ischemia down the brainstem.3 The rate of clinical deterioration, however, varies from patient to patient, may not occur in discernible steps or phases, or changes too quickly to be noted by observers. In general, the main features of the bedside neurological examination are: breathing pattern, motor function or responsiveness, assessment of the pupils, and ocular reflexes. The pattern of respiration is often impossible to evaluate because most comatose patients are initially intubated or mechanically ventilated. Furthermore, the correlation of specific lesions with certain breathing patterns is inexact.9 Cheyne-Stokes respiration, or crescendo/decrescendo tidal volumes alternating with apneas, may occur in patients with slowed circulation time from heart failure, or in systemically ill elderly patients, as well as those with bihemispheral cerebral lesions. Unrelenting hyperventilation is found more commonly with a primary pulmonary disorder (e.g., acute respiratory distress syndrome), and only rarely from an isolated midbrain lesion. Irregular, erratic breathing is typical of ataxic breathing, the precursor of respiratory arrest as the brainstem fails in rostral-caudal fashion to the level of the cardiorespiratory centers in the medulla.5 Spontaneous or stimulus-induced movements of the patient should be observed. Movement of a limb on command, or after a painful stimulus (sternal rub, nailbed compression, or rubbing the supraorbital ridge or angle of the jaw), is prognostically better than unresponsiveness. Spontaneous or stimulus-provoked decorticate posturing (unilateral or bilateral flexion of upper limbs with extension of lower limbs) occurs with dysfunction at the level of the cerebral hemispheres or diencephalon. Decerebrate posturing (unilateral or bilateral extension of upper and lower limbs) occurs with dysfunction at the level of the rednucleus (midbrain).5 Attempts to elicit the Babinski sign may lead to a “triple flexion” response, with flexion at the hip, knee, and ankle, also indicative of a corticospinal tract lesion.7 Asterixis may be observed bilaterally, with passive extension of the hands or feet, and, along with myoclonic jerks and tremulousness, suggests a metabolic cause of coma. Unilateral asterixis is seen with contralateral thalamic, midbrain, or parietal lobe lesions.10 Myoclonus or myoclonic jerks appear as sudden, shock-like muscle contractions of a limb or entire body, often triggered by tactile stimuli, and frequently seen with anoxic encephalopathy. Any other subtle, rhythmical, repetitive movements should be noted, such as twitching of the eyelid, face, or limb, or lateral gaze deviation with persistent nystagmus. These movements may be the only clinical signs of nonconvulsive or “electrical” status epilepticus, which may be the primary or major contributing cause of coma. Not all repetitive eye movements are epileptic, however. Ocular bobbing, a cyclical, fast jerk of both eyes downward, with a slower return to primary position, usually occurs with pontine lesions with poor prognosis.11 Any intubated, motionless, unresponsive patient with spontaneously blinking, opened eyes must be assessed for the de-efferented state of the “locked-in syndrome” (e.g., extensive pontine infarction from basilar artery occlusion). The examiner may be surprised to find that such a patient reliably answers yes-or-no questions by blinking once or twice. The examination of the pupils and the pupillary light reflex is a simple task, yet associated with a few pitfalls. Normal pupilloconstriction to a light stimulus involves efferent parasympathetic fibers with the third cranial nerve at the level of the midbrain. Thus, a lesion of the dorsal midbrain or third cranial nerve(s) produces larger, dilated, unreactive pupil(s). The sympathetic pupillodilator fibers leave the hypothalamus, descend the brainstem into the cervicothoracic spinal cord, exit into the sympathetic (stellate) ganglia, and ascend the carotid arteries to the orbit. Brainstem lesions caudal to the midbrain, therefore, disrupt these pupillodilator fibers, leaving tiny, pinpoint but reactive pupils. A comatose patient with pinpoint pupils will not always harbor a pontine lesion, however, because such pupils also occur with a narcotic drug overdose or in glaucoma patients using cholinergic (pilocarpine) eyedrops. Coma-producing lesions elsewhere tend to cause somewhat small, but reactive pupils, which are the usual finding in conscious elderly patients. Metabolic causes of coma also produce small pupils of equal size, with the light reflex preserved even after loss of other cranial nerve or brainstem reflexes (corneal, oculocephalic, oculovestibular, and gag reflexes).12 The finding of an enlarged, dilated, nonreactive pupil in an unresponsive patient is an ominous finding, generally representing uncal herniation from an ipsilateral mass. In such unconscious patients, computed tomography (CT) and magnetic resonance imaging (MRI) studies reveal that coma is produced by severe horizontal compression and shift of the diencephalon, which precedes obscuration of the perimesencephalic cisterns and pressure on the uncus itself.13,14 Horizontal shift and distortion of the upper brainstem, containing the ARAS, more readily creates coma than similar degrees of vertical displacement of the brainstem, such as that occurring with intracranial hypotension after lumbar puncture. Horizontal pineal displacement of 3 to 4 mm correlates with drowsiness, 6 to 8 mm with stupor, and over 8 mm with coma.13 The oculocephalic and oculovestibular reflexes (Fig. 1-1) are both normal brainstem reflexes, elicited and observed in the absence, or near absence, of cortical influence or control. Provided a cervical spine injury is absent or excluded, gentle, passive rotation of the patient’s head toward the left normally produces conjugate lateral rolling of both eyes to the right, and vice versa. Put another way, during lateral head rotation, the patient’s eyes tend to keep looking at the examiner when observing the patient “face to face.” This is the oculocephalic reflex or “doll’s eyes” maneuver. The oculovestibular, or “cold caloric” reflex, may be preserved or persist after the oculocephalic reflex is absent. With the patient’s head elevated ~30 degrees, ice water irrigation of the ear canal normally produces some turbulence or movement of endolymphatic fluid within the labyrinthine semicircular canals, causing a slow, tonic deviation of both eyes toward the irrigated ear. Lateral nystagmus, with rapid component to the opposite ear, requires some cortical function, and is thus not usually observed in the comatose subject. Care should be taken to ensure against a false negative response, instilling at least 50 cc of ice water for an adequate test stimulus. If cerumen or debris occludes the ear canal, a normal response may be prohibited. Water should not be instilled into an ear canal with ruptured tympanic membrane, because of the risk of infection.
Causes of Coma
Clinical Evaluation of Comatose Patients
Assessment of Acute Loss of Consciousness
Only gold members can continue reading. Log In or Register to continue







Full access? Get Clinical Tree


