Bone Diseases and the Central Nervous System
Roger N. Rosenberg
Alison M. Pack
PAGET DISEASE
This chronic disease of the adult skeleton, formerly known as osteitis deformans, is characterized by bowing and irregular flattening of the bones. Any or all skeletal bones may be affected, but the tibia, skull, and pelvis are the most frequent sites. Usually, multiple bones are affected, but in 15%, only one is involved. The disease is symptomatic in 15% to 20% of affected individuals. Pain is the most common symptom. Except for the skeletal deformities and pain, the disease causes disability only when the skull or spine is involved.
EPIDEMIOLOGY
There is a postmortem incidence of 3% in patients older than 40 years of age. Men and women are equally affected. The most common age at onset is in the fourth to sixth decades; it is rare before age 30 years. Genetic and environmental factors such as paramyxovirus infection and dietary deficiency of calcium may influence the presentation.
PATHOBIOLOGY
In affected bones, there is an imbalance between the formation and the resorption of bone. In most cases, there is a mixture of excessive bone formation and bone destruction. The areas of bone destruction are filled with hyperplastic vascular connective tissue. New bone formation may occur in the destroyed areas in an irregular disorganized manner. The metabolic disturbance is unknown.
CLINICAL FEATURES
Two types of neurologic symptoms appear: those owing to the abnormalities in bone and those owing to arteriosclerosis, a common accompaniment. The cerebral manifestations that occur with arteriosclerosis are identical to those seen in patients with arteriosclerosis in the absence of Paget disease.
The neurologic defects of osteitis deformans are usually related to pressure on the central nervous system (CNS) or nerve roots by the overgrowth of bone. Convulsive seizures, generalized or neuralgic head pain, cranial nerve palsies, and paraplegia occur in a few cases. Deafness caused by pressure on the auditory nerves is the most common symptom; unilateral facial palsy is the next most common. Loss of vision in one eye, visual field defects, or exophthalmos may occur when the sphenoid bone is affected. Compression of the spinal cord is more common than compression of the brain, which is extremely rare except when there is sarcomatous degeneration of the lesions. Platybasia may occur in advanced cases. Paget disease has also been described in a patient with basilar impression and Arnold-Chiari type I malformation.
DIAGNOSIS
The diagnosis of Paget disease is made from the patient’s appearance and the characteristic radiographic changes. Involvement of the skull in advanced cases is manifested by a generalized enlargement of the calvarium, anteroflexion of the head, and depression of the chin on the chest. When the spine is involved, the patient’s stature is shortened, the spine is flexed forward, and spinal mobility is greatly reduced.
Radiographically, the skull shows areas of increased bone density with loss of normal architecture, mingled with areas in which the density of the bone is decreased (Fig. 122.1). The margins of the bones are fuzzy and indistinct. The general appearance is that of an enormous skull with the bones of the vault covered with cotton wool. In advanced cases, there may be a flattening of the base of the skull on the cervical vertebrae (platybasia) with signs of damage to the lower cranial nerves, medulla, or cerebellum. Both computed tomography (CT) and magnetic resonance imaging (MRI) aid diagnosis.
The serum calcium content is normal, and the serum phosphorus is normal or only slightly increased. Serum alkaline phosphatase activity is increased; the level varies with the extent and activity of the process. It may be only slightly elevated when the disease is localized to one or two bones.
Diagnosis may be difficult if the clinical symptoms are mainly neurologic. In these instances, radiographs of the pelvis and legs or a general survey of the entire skeleton may establish the diagnosis. Rarely, it may be impossible to distinguish monophasic Paget disease of the skull from osteoblastic metastases. Search for a primary neoplasm, particularly in the prostate, or biopsy of one of the lesions in the skull may be necessary in those cases.
TREATMENT
Bisphosphonates such as pamidronate, alendronate, risedronate, and zoledronic acid are considered the treatment of choice. These agents inhibit osteoblastic bone resorption. They have been shown to heal radiologic lesions and restore normal histology. The longterm effects of these drugs on disease progression have not been adequately studied. The most common indication is for pain. Less potent second-line choices include etidronate and tiludronate. Calcium and vitamin D repletion is necessary to avoid hypocalcemia secondary to bisphosphonates. The value of these medical therapies can be evaluated by reduction of serum levels of alkaline phosphatase measured at 4-month intervals and annual radiographs of specific lesions. Decompression of the spinal cord may be indicated for myelopathy secondary to stenosis created by the enlarged vertebrae. Similarly, platybasia may require to decompression of the posterior fossa to relieve cranial nerve dysfunction.
Calcitonin may also be given to inhibit the osteolytic process. Salmon calcitonin is given in subcutaneous injections of 50 to 100 units daily. Improvement of osteolytic lesions and reversal of neurologic manifestations have been noted with long-term therapy. About 25% of the patients develop serum antibodies to salmon calcitonin, sometimes in titers high enough to make the person resistant to the hormonal action of calcitonin; under these circumstances, human calcitonin may be effective.
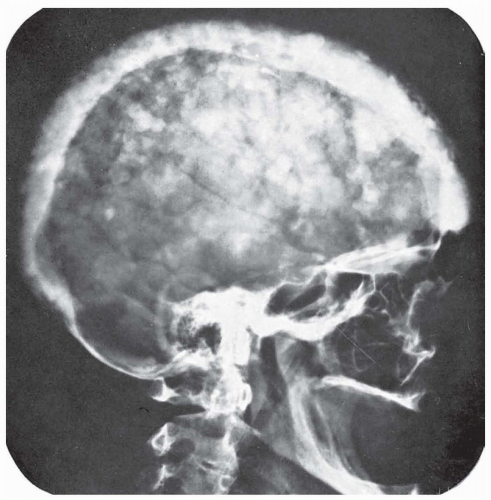 FIGURE 122.1 Osteitis deformans (Paget disease) of the skull. (Courtesy of Dr. Juan Taveras.) |
OUTCOME
The course is variable but usually extends for decades. The neurologic lesions seldom lead to serious disability other than deafness, convulsive seizures, or compression of the spinal cord.
FIBROUS DYSPLASIA
The skull and the bones in other parts of the body are occasionally involved by a process characterized by small areas of bone destruction or massive sclerotic overgrowth. The clinical picture of fibrous dysplasia is related to the site and extent of the bone overgrowth. Fibrous dysplasia can involve one (monostotic form) or multiple (polyostotic form) bones. Both types may be associated with hyperfunctional endocrinopathies and café au lait lesions referred to as the McCune-Albright syndrome. Fibrous dysplasia and McCune-Albright syndrome are caused by a sporadic postzygotic missense mutation in the somatic cells of the GNAS complex locus located on chromosome 20q13. Overall, this leads to defects in osteoblast differentiation and proliferation of osteoblast progenitors leading to abnormal bone. The bones of the skull most often affected are the frontal (56%) and sphenoid (48%) bones. A recent Danish case series of 26 subjects revealed that 80% had involvement of craniofacial bones. Diffuse involvement of the entire skull produces leontiasis ossea, with exophthalmos, optic atrophy, and cranial nerve palsies (Fig. 122.2).
In addition to the disfiguration of the skull in the polyostotic form, symptoms of the monostotic form of the disease include headache, convulsions, exophthalmos, optic atrophy, and deafness. Most patients are, however, asymptomatic. Symptoms may begin at any age, but onset usually occurs in early adult life. The family history is negative, and there is no racial or sexual predominance.
Treatment is limited. Surgical intervention can be used to treat fracture and bony deformities. Although not well studied, bisphosphonates have been used to treat bone pain. Results are mixed and no randomized clinical trial has been done.
ACHONDROPLASIA
Achondroplasia (chondrodystrophy) is the most frequent form of skeletal dysplasia causing dwarfism. It is characterized by short arms and legs, lumbar lordosis, and enlargement of the head caused by mutations in the fibroblast growth factor receptor 3 gene (FGFR3). The gene product is expressed in cartilage. A frequent FGFR3 mutation is a Gl 138A codon mutation with GGG to AGG or CGG substitutions, resulting in an exchange of glycine at position 380 in the FGFR3 protein to arginine. This mutation results in a gain of negative function, producing an inactive FGFR and resultant dwarfism. The disease is rare and is estimated to occur in 15 of 1 million births in the United States. It is inherited as an autosomal dominant trait, although 80% of cases are sporadic.
Symptoms of involvement of the nervous system sometimes develop as a result of hydrocephalus, compression of the medulla and cervical cord at the level of the foramen magnum, compression of the spinal cord by ruptured intervertebral disk, and bone
compression of the lower thoracic or lumbar cord. Seizures, ataxia, and paraplegia are the most common symptoms. Mental development is usually normal. Mortality rates are significantly elevated secondary to accidental, neurologic, and heart diseaserelated causes.
compression of the lower thoracic or lumbar cord. Seizures, ataxia, and paraplegia are the most common symptoms. Mental development is usually normal. Mortality rates are significantly elevated secondary to accidental, neurologic, and heart diseaserelated causes.
The diagnosis is made from the characteristic body configuration of short arms and legs, normal-sized trunk, enlargement of the head, and changes in the radiographs of the skeleton. Previously, CT was used to evaluate neurologic complications of achondroplasia. Currently, MRI of the brain and spine best define the presence of hydrocephalus and degree of ventricular compensation as well as whether cord compression is present. Somatosensoryevoked potential may be useful for detecting cervical myelopathy. Many affected infants die in the perinatal period, although a normal life span is possible for patients with less severe involvement of the bones.
Shunting procedures may be needed for hydrocephalus caused by involvement of the bones at the base of the skull. Laminectomy is indicated for signs of cord compression. The mutations in the FGFR3 gene at 4p are usually new mutations and result in autosomal dominant inheritance.
ANKYLOSING SPONDYLITIS
Ankylosing spondylitis is considered the prototype of the spondyloarthropathies. It typically occurs in younger persons and men are affected two to three times more commonly than women. Its etiology is likely secondary to multiple genetic and environmental factors including a strong association with the gene for human leukocyte antigen (HLA)-B27. The condition is common, affecting an estimated 1.4% of the general population. Neurologic symptoms have been considered to be uncommon but have only received limited study. A recent study investigated this question further among 24 patients recruited from an Egyptian outpatient rheumatologic clinic compared to age and sex matched controls. Neurologic manifestations occurred in 25% of the patients; 8.3% had myelopathy and 16.7% had radiculopathy. Subclinical neurologic complications as evident by neurophysiologic test abnormalities were common, as over 70% had at least one abnormal finding.
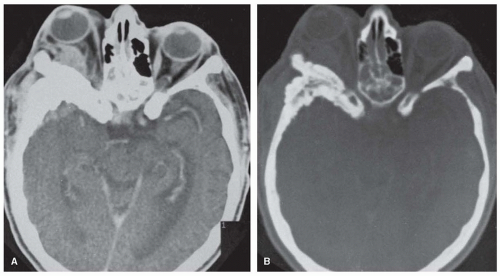 FIGURE 122.2 Fibrous dysplasia. CT. A: Axial contrast-enhanced scan shows proptosis on right with abnormal soft-tissue enhancement within orbit and middle cranial fossa. B: Bone window depicts pronounced thickening of sphenoid bone. (Courtesy of Dr. T. L. Chi.) |
This inflammatory disorder affects ligamentous insertions into bones; at first, it usually affects the sacroiliac joints and lumbar spine. In some patients, the entire spine is involved, with ossification of the ligaments and fusion of the vertebra. The spine becomes rigid and susceptible to a variety of disorders that may affect the spinal cord, including fractures and dislocations, atlanto-occipital dislocation, and spinal stenosis.
A cauda equina syndrome may appear in patients with longstanding spondylitis. Signs and symptoms are symmetric, with weakness, wasting, and sensory loss in lumbosacral myotomes. Autonomic dysfunction and more specifically parasympathetic dysfunction may occur. Bladder and bowel are commonly affected, and pain may be severe. The mechanism is not clear. Although concomitant arachnoiditis has been suspected as the cause, the syndrome appears late, when there is little evidence that the underlying spondylitis is active. Moreover, there is little inflammation at postmortem examination, which is likely to show chronic fibrosis. There is erosion of posterior bone elements, and, in earlier days, myelography showed enlargement of the caudal sac and prominent diverticulae of the arachnoid. CT shows similar pathology, but MRI is more illuminating, showing nerve root thickening and sometimes enhancement of dura and nerve roots; that pattern suggests inflammation of the arachnoid structures, supporting the earlier theory. Surgery is generally ineffective and has sometimes been deleterious, although there have been rare reports of some relief. Steroid therapy has been similarly without benefit. Physical therapy may be useful. Studies suggest the use of nonsteroidal anti-inflammatory drugs (NSAIDs) or selective cyclo-oxygenase-2 inhibitors for symptomatic patients. Tumor necrosis factor (TNF) inhibitors are useful for patients whose symptoms persist despite treatment with NSAIDs.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


