Chapter 52B Cancer and the Nervous System
Pathology and Molecular Genetics
General Principles of Nervous System Tumor Biology
Nervous system tumors, like other human neoplasms, are clonal proliferations that develop secondary to changes in key growth regulatory genes. Such genes fall into several different classes: growth-promoting oncogenes that abnormally activate in tumors, growth-checking tumor suppressor genes that inactivate in tumors, cell death genes that are impaired in tumors, and deoxyribonucleic acid (DNA) repair genes that deregulate in tumors. In combination, these genetic changes result in powerful growth advantages that enable cells to proliferate, evolve, and disseminate (Louis, 2006).
Some of the particular genes affected in human nervous system tumors are newly discovered and are discussed with the respective tumor type. Interestingly, most of the identified genes are deregulated in many other types of human cancers (a notable exception being IDH1), and the manner in which such deregulation specifically affects nervous system cells remains unclear. Certain nervous system cells are probably more susceptible to neoplastic transformation than others. For example, the far greater frequency of neoplastic transformation of gliomas compared to neuronal tumors is attributable to the greater neoplastic vulnerability of glial cells. Because oncogenic transformation requires cell division, postmitotic cells such as neurons should not be susceptible to tumorigenic events. However, it remains possible that nervous system tumors arise when oncogenic changes occur in precursor cells rather than mature glial cells or neurons. Such precursor neural stem cells reside in the brain into adult life (Vescovi et al., 2006). In this sense, the greater frequency of gliomas may relate to the particular paths of differentiation that are followed after specific tumorigenic genetic changes (i.e., transformed neuroepithelial progenitors may have to undergo specific molecular events to become tumors, with these same events essentially restricting cells to glial differentiation).
It is unknown why nervous system cells rather than other cells in the body transform in these instances because the etiologies of brain tumors remain unknown. To date, radiation and hereditary predisposition are the only clearly implicated factors in the genesis of nervous system tumors. See Chapter 52A for a discussion of ionizing radiation as a risk factor for specific types of brain tumors. Hereditary predisposition to brain tumors is rare and confined to specific syndromes (e.g., neurofibromatosis type 1 [NF1], neurofibromatosis type 2 [NF2], tuberous sclerosis, von Hippel-Lindau disease, Li-Fraumeni syndrome, Turcot syndrome, and Gorlin syndrome). These conditions highlight the critical roles played by specific genes in brain tumorigenesis.
History of Nervous System Tumor Classification Schemes
The foundation of the first comprehensive classification of nervous system tumors, formulated by Percival Bailey and Harvey Cushing in 1926, presumed parallels between embryological and neoplastic cells. In large part, this histogenetic “cell of origin” model still forms the basis for current nomenclature; renewed interest in the role of developmental pathways in tumorigenesis has emerged in recent years. In 1949, as a means of enhancing the clinical utility of tumor classification, Kernohan contributed a tumor grading system whose purpose was assessing patient prognosis. Russell and Rubinstein modified and updated the Bailey and Cushing system during the 1960s, 70s, and 80s. Further updates were incorporated into the World Health Organization (WHO) classification, first completed in 1979 and then revised in 1993 and 2007 (Louis et al., 2007). The WHO classification system has become the system most widely used by neuropathologists today.
The WHO classification currently lists more than 100 types of nervous system tumors and their variants (Louis et al., 2007). This level of complexity may seem daunting at first, but consideration of key clinical and imaging characteristics typically narrows the differential diagnosis to only a few common possibilities (Table 52B.1). For example, the differential diagnosis varies substantially for supratentorial versus infratentorial, pediatric versus adult, and enhancing versus nonenhancing tumors.
Table 52B.1 Common Central Nervous System Tumor Diagnoses by Location, Age, and Imaging Characteristics
| Location | Child/Young Adult | Older Adult |
|---|---|---|
| Cerebral/supratentorial | ||
| Cerebellar/infratentorial | ||
| Brainstem | ||
| Spinal cord (intraaxial) | ||
| Extraaxial/dural | ||
| Intrasellar | ||
| Suprasellar/hypothalamic/optic pathway/third ventricle | ||
| Pineal | ||
| Thalamus | ||
| Cerebellopontine angle | ||
| Lateral ventricle | ||
| Nerve root/paraspinal |
AT/RT, Atypical teratoid/rhabdoid tumor; DNT, dysembryoplastic neuroepithelial tumor; E, enhancing; MEN, mural enhancing nodule; MPNST, malignant peripheral nerve sheath tumor; NE, nonenhancing; NF1, neurofibromatosis type 1; NF2, neurofibromatosis type 2; PNET, primitive neuroectodermal tumor, SEGA, subependymal giant cell astrocytoma.
General Histopathological Features and Techniques
General Histopathological Features
Palisading
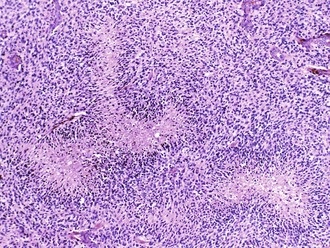
Palisading, or lining up of tumor cells, is characteristic of schwannomas, in which collections of palisaded nuclei are termed Verocay bodies. The term describes a collection of neoplastic cells arranged in parallel arrays, or palisades. In glioblastomas, palisading (also known as pseudopalisades) occurs around necrosis, with serpiginous zones of necrosis lined by hypercellular viable tumor nuclei (Fig. 52B.1).
Rosettes
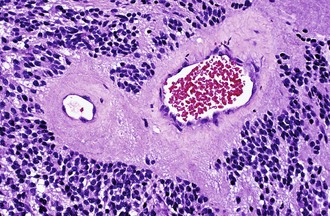
Characteristic of perivascular rosettes is a peripheral ring of tumor nuclei surrounding a central blood vessel with a nuclear-free eosinophilic zone in between (Fig. 52B.2). The nuclear-free zone derives from tapering cellular processes that radiate from the tumor cells to the vessel. Perivascular rosettes are most typical of ependymomas, and glial fibrillary acidic protein (GFAP) immunostains highlight the perivascular processes. Occasionally, similar axon-bearing structures appear in neuronal tumors as well (e.g., neurocytomas, pineocytomas, various forms of PNET).
Microvascular Proliferation
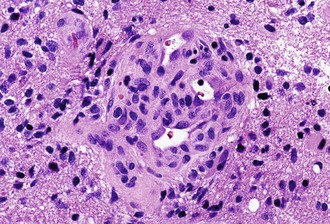
Microvascular (endothelial and pericytic/smooth muscle cell) proliferation represents an important and interesting response to tumor-associated and hypoxia-associated angiogenesis factors. It occurs in either benign or malignant CNS neoplasms, although it is more common in the malignant. In pilocytic astrocytoma, the new vessels may take on a delicate glomeruloid or “wicker-works” configuration, usually with a single-layered flat endothelial lining. In malignant tumors such as glioblastoma, these vessels are also glomeruloid but additionally display multilayering, with enlarged or “hyperplastic” endothelial cells (Fig. 52B.3). In either case, these neovascular structures lack the tight junctions normally contributing to the blood-brain barrier. Therefore, they typically leak intravascular contrast material, thus accounting for the tumor-associated enhancement detected radiologically.
Frozen Sections and Touch Imprints/Smears
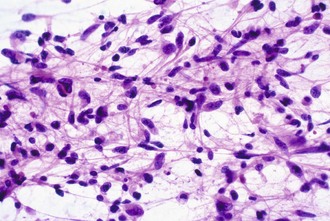
Intraoperative frozen sections are more difficult to interpret than fixed tissue sections, but their use has the advantage of speed, requiring only a few minutes to prepare. This technique is requested for several reasons, including simple curiosity and a desire to provide rapid feedback to patients and their families. However, the most important reasons for performing this technique are to ensure that the pathologist obtains a representative sample for permanent sections and to provide information that may alter the surgical procedure. For example, a neurosurgeon may opt to stop at a limited biopsy for a suspected lymphoma, aggressively resect for an ependymoma, or send additional material to the microbiology laboratory for an abscess. Because the process of tissue freezing produces significant cytological artifacts, another popular technique has been the preparation of touch imprints, or smears, from fresh tissue. Although the underlying architecture is lost, the tissue requirements are minimal, and the cytological preservation is excellent (Fig. 52B.4). This may be critical for diagnoses based primarily on nuclear cytology, such as reactive gliosis versus low-grade diffuse glioma. It is also important to realize that the frozen section technique results in substantial tissue loss and morphological artifacts in the thawed residual specimen. Therefore, it is best to omit its use in limited biopsies for which the surgical procedure will not be altered. In other words, it is imperative to save at least some optimally fixed nonfrozen tissue for final diagnosis on sections from the fixed embedded material.
Molecular Diagnostics
Few molecular diagnostic tests have become routinely used or standard of care in neuro-oncology. The most notable and most widely used is chromosome 1p and 19q testing as a prognostic/management tool for patients with oligodendroglial tumors. FISH has the advantage of simplicity, morphological preservation, minimal tissue and purity requirements, and a lack of necessity for microdissection or matching blood/nonneoplastic tissue. However, it requires some experience for accurate interpretation, especially in cases with aneuploid populations of tumor cells. It also utilizes large probes (100–300 kb) and is insensitive to very small deletions. In selected cases, FISH has been used to evaluate epidermal growth factor receptor (EGFR) amplification to distinguish small cell glioblastoma from anaplastic oligodendroglioma. It is likely that many more applications of molecular diagnostics will become incorporated into diagnostic neuropathology laboratories in the near future, but it is also important to note that specific immunohistochemical assays may “substitute” for molecular diagnostic approaches in certain situations. For example, immunohistochemical absence of the product of the INI1 gene on chromosome 22q is now widely accepted as a sensitive and specific marker for atypical teratoid/rhabdoid tumors (AT/RTs) (Judkins et al., 2004). Similarly, because IDH1 is a frequently mutated gene in lower-grade gliomas and secondary glioblastoma (Yan et al., 2009), antibodies to the most common mutant form of the IDH1 protein have demonstrated clinical utility in the pathological assessment of infiltrating glioma (Camelo-Piragua et al., 2009).
Primary Neuroepithelial Tumors
Diffuse Astrocytoma (WHO Grade II)
Microscopically, a slight to moderate increase in cellularity occurs, and the primary distinction from reactive astrocytosis rests on the finding of cytological atypia. Diffuse astrocytoma cells can take on various morphological appearances, including fibrillary, gemistocytic, and protoplasmic types. Fibrillary morphology is most common and consists of irregular elongated hyperchromatic nuclei, either appearing “naked” in an otherwise fibrillary background or displaying discernible cytoplasmic processes (see Fig. 52B.4). GFAP immunostains highlight the latter, although this is neither absolutely sensitive nor specific, because (1) some astrocytomas harbor minimal quantities of GFAP-positive cytoplasm, (2) interpretation may be difficult owing to staining of nonneoplastic astrocytic elements or high background in general, and (3) other gliomas display GFAP immunoreactivity. By definition, grade II astrocytomas lack mitotic activity, microvascular proliferation, and necrosis. Ancillary staining for MIB-1 (Ki-67) generally reveals a low proliferative index as well.
Recently, a large proportion of low-grade gliomas (including 90% of grade II astrocytomas) were found to have mutations in IDH1 and IDH2, even though the mutation was initially described in 12% of (largely secondary) glioblastomas (Parsons et al., 2008). IDH1 encodes isocitrate dehydrogenase, a component of the citric acid cycle. The role of IDH1 in glioma genesis remains uncertain, although recent work suggests that this loss of function mutation may cause accumulation of substrate, triggering HIF signaling pathways (Zhao et al., 2009). Others have proposed that tumorigenesis may be a consequence of 2-hydroxyglutarate formation (Dang et al., 2009). IDH1 mutations in low-grade glioma are tightly correlated with earlier age at diagnosis and tumor grade, appear to confer prognostic benefit beyond tumor grade, and are frequently associated with O6-methylguanine-DNA methyltransferase (MGMT) methylation (Sanson et al., 2009).
Chromosome 17p losses and mutations of TP53 are common molecular genetic events in diffuse astrocytomas, seen in approximately half of cases (Louis, 2006). Those with gene mutation usually display strong nuclear immunostaining for p53 protein, although the association is imperfect, since the protein is stabilizable by other mechanisms, and many immunopositive cases lack TP53 mutations. Those cases that evolve to glioblastoma (secondary glioblastoma) typically retain evidence of TP53 mutation and may even show additional TP53 gene mutations or clonal expansion of the tumor cells with mutation. On the other hand, de novo, or primary, glioblastomas rarely display such mutations. Other common alterations involve the overexpression of platelet-derived growth factor receptor alpha (PDGFα) in astrocytomas of all grades and losses of chromosome 22.
Anaplastic Astrocytoma (WHO Grade III)
Histologically, anaplastic astrocytomas are more cellular than grade II astrocytomas and display a greater degree of proliferation. The presence of mitotic figures primarily defines these tumors. One astrocytoma variant that commonly presents at the anaplastic or grade III level is the gemistocytic (“stuffed cell”) astrocytoma. Characterized by strongly GFAP-positive cells with eccentric bellies of eosinophilic cytoplasm, this tumor type has a high incidence of progression to glioblastoma. Interestingly, the small-cell astrocytoma elements in the background primarily constitute the proliferating element. Another feature that may help distinguish anaplastic astrocytomas from grade II astrocytomas is the generally higher Ki-67 or MIB-1 proliferative index on immunostains (Giannini et al., 1999a).
Anaplastic astrocytomas share the high frequency of TP53, IDH1, and IDH2 mutations seen in grade II astrocytomas. Inactivation of the retinoblastoma protein (Rb) cell-cycle regulatory pathway is also common and may be primarily responsible for the increased proliferation observed histologically. Homozygous deletion of the CDKN2A/p16 gene on 9p is the most common mechanism for disabling this pathway, although deletions of the RB gene and CDK4 amplifications are alternative aberrations (Louis, 2006). Amplifications of the EGFR gene and losses of PTEN on 10q occur less frequently in anaplastic astrocytomas than in glioblastomas and are virtually absent in tumors containing an IDH1 mutation (Sanson et al., 2009). When present, EGFR amplification may suggest either a worse prognosis or the possibility of undersampling/undergrading in a glioblastoma (Smith et al., 2001).
Glioblastoma (WHO Grade IV)
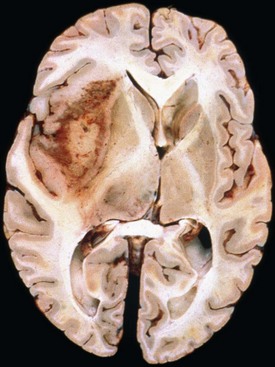
Glioblastoma most commonly occurs in the deep white matter, basal ganglia, or thalamus and is rarely found in the cerebellum or spinal cord. The gross and microscopic appearance is heterogeneous. Replacing the affected portion of the brain is a single mass that grossly may appear deceptively well circumscribed but microscopically infiltrates widely, often spreading to the opposite hemisphere via the corpus callosum (butterfly lesion). Multifocal tumors may occur and in most cases likely represent separate regions of malignant transformation within a widely disseminated lower-grade astrocytoma such as gliomatosis cerebri. In advanced stages, the tumor may extend into the meninges or the ventricle. Seeding of the neuraxis as multiple implants on the brain or ventricular surfaces is an atypical growth pattern, and extracranial metastases are extremely rare. The cut surface has a variegated appearance (Fig. 52B.5) characterized by central yellow or white zones of necrosis and hemorrhage surrounded by a hyperemic ring (endothelial hyperplasia) and “edematous” brain with variable mixtures of vasogenic edema, gliosis, and tumor infiltrates. Microscopically characterizing glioblastomas are all the features of anaplastic astrocytoma plus endothelial hyperplasia or necrosis. The endothelial hyperplasia is thickened or glomeruloid vessels with multilayering (see Fig. 52B.3), representing a form of tumor-induced angiogenesis, a potential target for novel therapies. The necrosis is often associated with a characteristic serpiginous distribution and associated nuclear pseudopalisading (see Fig. 52B.1). Several histological variants exist, including giant cell glioblastoma, small cell glioblastoma, and gliosarcoma. Characteristic of the latter is a sarcomatous element, currently felt to arise most likely from mesenchymal metaplasia within a glioblastoma. No significant clinical differences are identifiable in these variants when compared with conventional glioblastoma, although gliosarcomas more frequently occur in the temporal lobes.
Glioblastomas are among the best genetically characterized CNS tumors and often harbor numerous cytogenetic and molecular genetic aberrations (Louis, 2006). Mutations of TP53, IDH1, and IDH2 are less common than in diffuse astrocytomas or anaplastic astrocytomas and occur more often in secondary glioblastomas. Arising in younger patients after malignant transformation of a lower-grade precursor, TP53 mutations occur at greater frequency in giant cell glioblastomas (Louis et al., 2001). In contrast, 30% to 40% of glioblastomas harbor EGFR amplifications, often with an associated constitutively activating mutation, and these occur mainly in the primary, or de novo, glioblastoma (Louis et al., 2001). Defining the latter is the lack of a precursor lesion, typically in an older patient with rapid clinical onset. EGFR amplifications are also particularly common (≈70%) in the small-cell variant. These tumors are composed of small round cells with minimal cytoplasm and a remarkably brisk mitotic index. Owing to the rounded, uniform nuclei, a misdiagnosis of anaplastic oligodendrogliomas may occur, although small cell glioblastoma share the demographic features and poor prognosis of glioblastomas in general and do not harbor the 1p and 19q deletions seen in oligodendrogliomas. Alterations of the p16/CDK4/RB pathway are nearly universal in glioblastoma, and many cases harbor chromosome 10 losses (mostly primary glioblastomas). The PTEN gene located on chromosome 10q23 mutates in a subset of these and may have prognostic significance in certain patient populations. Additional tumor suppressor genes on both the long and short arms of chromosome 10 are suspected in the remaining group of PTEN wild-type tumors with demonstrable LOH. The list of additional glioblastoma-associated alterations is growing rapidly. Recent large-scale sequencing studies have shown that glioblastomas have somatic mutations of multiple genes (Parsons et al., 2008; TCGA Network, 2008). The studies have confirmed our knowledge of the incidence of mutated and/or amplified genes such as EGFR, PTEN, RB1, and PIK3CA and showed higher prevalence for mutations in TP53, NF1, ERBB2, and PIK3R1 (TCGA Network, 2008) as well as new candidate genes involved in tumorigenesis, such as IDH1 (Parsons et al., 2008). Nevertheless, the focus of intensive research is on mechanisms of invasion, cellular migration, proliferation, angiogenesis, apoptosis deregulation, and therapeutic resistance (Louis, 2006).
Early evidence suggests that molecular analysis of glioblastomas may have prognostic and predictive relevance. For example, methylation of the MGMT gene promoter, leading to down-regulation of MGMT expression, appears prognostic of improved survival in patients with glioblastoma treated with the alkylating agent, temozolomide (Hegi et al., 2005). However, emerging data suggest that prognostic benefit associated with MGMT methylation in glioblastoma may be unrelated to the mode of therapy administered (Rivera et al., 2010). Furthermore, resistance to alkylating chemotherapy in glioblastoma is associated with therapy-related inactivation of MSH6, a DNA mismatch repair enzyme, allowing increased mutagenesis and the emergence of a hypermutated phenotype (Cahill et al., 2007; Yip et al., 2009).
Molecular approaches to global-expression profiling potentially provide powerful means to distinguish glioblastomas from other malignant gliomas that follow distinct clinical courses (Nutt et al., 2003). Moreover, gene-expression profiling of glioblastoma has progressed from augmenting information provided by histological examination to characterization of molecular subtypes based on different gene expression patterns in each group (Sulman et al., 2009). For example, The Cancer Genome Atlas (TCGA) Research Network published a molecular classification of glioblastoma based on expression of signature genes (Verhaak et al., 2010). Of the four subgroups identified (named based on prior work and genes expressed as proneural, neural, mesenchymal, and classic subtypes), the proneural group showed distinctive clinical characteristics including overrepresentation among younger patients, mutations in IDH1, and prior or concomitant areas with low-grade histology—indicating that these are secondary glioblastomas (Verhaak et al., 2010). There also appears to be a CpG island methylator phenotype (CIMP) of glioma belonging to the same proneural molecular subgroup that is common among low-grade gliomas, with a high proportion showing mutations in IDH1 (Noushmehr et al., 2010). While perhaps allowing more coherent selection of potential targets for molecular therapy and validating the role of surrogate markers such as IDH1 for identifying prognostically significant subgroups of glioma, molecular profiling remains largely investigative to date.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


