4 Most malignant tumors are thought to arise from a series of genetic alterations that result in the loss of control over cell proliferation and the ability to eliminate aberrant cells through the normal pathway of programmed cell death (apoptosis). Both cytogenetics and molecular biology have contributed to our understanding of the nonrandom genetic changes that contribute to the malignant phenotype and its progressive nature. Many different neoplasms share some genetic changes, whereas others are restricted to specific tumor types. With the completion of the human genome project and the development of new technology, progress is being made in characterizing these genetic lesions. In many tumors specific aberrations are now associated with the tumor type and grade, and they have also begun to distinguish subsets within a tumor. Equally important is the potential that each deoxyribonucleic acid (DNA) lesion represents for the design of new patient therapies. Before the cytogenetic and molecular findings for central nervous system (CNS) spinal cord tumors and peripheral nerve tumors are described in this chapter, the cell cycle and apoptosis pathways are reviewed briefly. The multistep process of transforming a normal cell, either in vitro or in vivo, requires a series of changes. Many of the changes involve two families of genes referred to as oncogenes and tumor-suppressor genes. Humans possess DNA sequences (genes) similar to viral oncogenes (genes that are capable of transforming normal cells). When these genes are functioning normally, they are called proto-oncogenes. Proto-oncogenes encode several types of proteins. Some of these proteins carry messages from the cell’s surface to the nucleus (signal transduction), where they initiate an effect. Others stimulate a cell to divide (growth factors), and still others are receptors that transmit a message through the cell membrane from the external environment when stimulated (Table 4-1). Proto-oncogenes become oncogenes when they are activated by a mutation or when the gene copy number increases due to a gene duplication or aberrant chromosome segregation (gene amplification). Oncogenes act in a dominant manner. Only one allele in the pair needs to be altered for aberrant expression to occur. Tumor-suppressor genes function exactly the opposite of oncogenes (Table 4-2). Their function is to slow or suppress growth entirely. Tumor-suppressor genes can be inactivated by a mutation or silenced by the process of methylation. These genes also can be lost through chromosomal segregation errors, chromosomal deletions, or genetic recombination. Mitotic figures are one of the hallmarks of malignancy in tumors; however, the signals that initiate the commitment of a tumor cell to divide occur well before the presence of a mitotic figure. The cell cycle consists of four phases: G1, S, G2, and M (Fig. 4-1). Each phase in the cell cycle is controlled by several families of proteins that can either activate or inhibit the progression of events in this pathway. In this capacity the aberrant expression of oncogenes and tumor-suppressor genes releases cells from the normal constraints controlling cell proliferation.
Cellular and Molecular Biology of Central Nervous System Spinal and Peripheral Nerve Neoplasms
 Cell Cycle
Cell Cycle
| Oncogenes | Chromosome location |
| MDM2 | 12q |
| Gli | 12q13–14.3 |
| MET | 7Q31 |
| MYC | 8q24 |
| abl | 9q34 |
| erbA | 17p11–q21 |
| fos | 2q21–q31 |
| H-ras | 11p |
| K-ras | 6q23–q12 |
| N-ras | 1p22–p31 |
| src | 20q12–q13 |
| 1p34-pter | |
| ros-1 | 6q16–q22 |
| c-sis | 22q12.3–13.1 |
| Growth factors | Chromosome location |
| PDGF A | 7p22 |
| PDGF B | 22q12–13.1 |
| PDGFR-α | 4q12 |
| PDGFR-β | 5 |
| EGFR | 7p12 |
| EGF | 4q25 |
| aFGF | 5q31.3–q33.2 |
| bFGF | 4q26 |
| TGF-α | 2p13 |
| TGF-β1 | 19q13.1 |
| TGF-β2 | 1q41 |
| TGF-β1 | 14q24 |
| TGFBR1 | 9q22 |
| TGFBR2 | 3p22 |
| TGFBR3 | 1p33–p32 |
| VEGF | 6p12 |
The cell cycle is an orderly sequence of phases (G0→G1→S→G2→M) that has checkpoints to determine if the cell should proceed to the next step. G0 cells such as neurons undergo terminal differentiation shortly after birth and do not reenter the cell cycle. Other quiescent cells that have not undergone terminal differentiation may enter the cell cycle upon receiving the appropriate signal. The first phase, G1, is characterized by cell growth and the synthesis of cellular proteins necessary for DNA synthesis. G1 is generally the longest phase in the cell cycle. In the S phase each DNA molecule is replicated or synthesized. Completing S phase each strand of DNA has made a copy of itself (sister chromatid strands) joined at a single region referred to as the centromere or kinetochore on a chromosome. This region is the last to replicate itself to permit separation of the chromosomes during the mitosis. Shorter than either the G1 or S phase is the G2 phase, the phase that determines if DNA replication is complete. Once replication is completed the cell enters the mitotic phase, which consists of prophase, metaphase, anaphase, and telophase. Normal glia cells are programmed to divide and do so to grow, repair, and replace cells.
| Tumor-suppressor gene | Chromosome location |
| RB | 13q14.2 |
| PTEN/MMAC1 | 10q23.3 |
| DCC | 18q21 |
| p16INK4αMTS1 | 9p21 |
| MTS2 | 9p21 |
| WAF1 | 6p22.2 |
| Chromosome 19q | |
| Chromosome 22q | |
| TP53 | 17p13.1 |
| TP73 | 1p36 |
| TP63 | 3q27–q29 |
When a cell is stimulated to divide, many proteins are involved in controlling the process. The cyclin-dependent kinases (CDKs) are holoenzymes that contain a regulatory (cyclin) and a catalytic (CDK) subunit. The cyclin family of proteins consists of cyclins A, B, D, and E, and they act in a relatively restricted area of the cell cycle. One of the D cyclins (D1, D2, and D3) joins with the catalytic proteins (CDK4 or CDK6) to yield six different CDK holoenzymes that function only in the G1 phase (Fig. 4-1). The cyclins are responsible for determining the timing of the activation of the CDKs and their substrate specificity.1 Additional protein families modulate the activity of the cyclins and CDKs by suppressing their function. These CDK inhibitors (CDKIs) are considered to be candidate tumor-suppressor genes because of their suppressive activity. Two major groups of CDKIs exist: the CIP/KIP or p21-like family (p21Cip1, p27Kip1, and p57Kip2) and the INK family or p16-like family (p16INK4a, p15INK4b, p18INK4c, and p19INK4d of genes; Fig. 4-1).
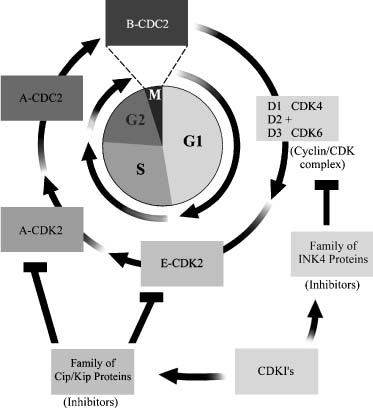
When the cell receives a mitotic signal, it initiates the formation of the cyclin/CDK complex that phosphorylates the Rb protein. Phosphorylation of the Rb protein eliminates its repressive action over the E2F proteins that initiate the transcription of genes (e.g., cyclin E and A) that permit the cell cycle to progress.2
 Programmed Cell Death (Apoptosis)
Programmed Cell Death (Apoptosis)
In contrast to the cell cycle, which promotes growth, the programmed cell death pathway is designed to destroy cells. Like the cell cycle, this pathway is also a genetically controlled cascade of cellular events that leads to the elimination of cells (Fig. 4-2). This program is designed to function when more cells are made than can be used or when cells become damaged and cannot be repaired properly. This determination is made at one of the checkpoints in the cell cycle. Thus, when ionizing or ultraviolet irradiation, cytotoxic drugs, mutagens, or carcinogens damage a cell, it must be repaired, or it is directed to the apoptotic pathway for elimination. The proteins that control the apoptotic pathway either suppress cell death or induce it.
The key proteins that promote cell survival are Bcl-2 and Bcl-xL. They suppress apoptosis by blocking the release of cytochrome c from mitochondria. If cytochrome c is released, the caspases/ICE family of proteins is activated and causes cytoplasmic and chromatin condensation and DNA fragmentation. The proteins that promote cell death by blocking the function of the Bcl-2 family of genes are named BAX and Bad and Bid. As in the cell cycle, phosphorylation and dephosphorylation of these proteins are critical events that mediate the activation or inhibition of this pathway.
Apoptosis is an important mechanism in animal development and in diseases such as cancer. When the genes controlling this pathway are deregulated, cells develop inappropriate survival and tumorigenic properties. One of the key genes in this decision-making process of whether cells survive or die is the TP53 gene, the same gene that is critical in the cell cycle (Fig. 4-1). This gene codes for the nuclear protein p53, which regulates whether cells survive or die following DNA damage caused by radio- or chemotherapy. The p53 protein is involved in at least two checkpoints in the cell cycle, the G1→S and the G2→M transitions (Fig. 4-1). These checkpoints are stages in the cell cycle that permit the repair of DNA damage in a cell before allowing it to proceed to the next phase. Which pathway is selected for a cell appears to be related to the cell type and the DNA damage incurred.
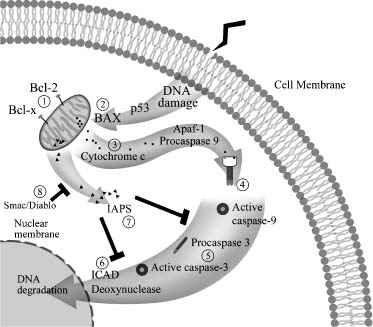
Therefore, p53 is of considerable importance in the treatment of cancer patients because chemotherapy drugs, such as 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU) and cisplatin, induce DNA damage that can activate the p53 apoptotic pathway and cause transformed cells to be eliminated. For such treatments to be effective, normal p53 protein must be present in the cell. Unfortunately, about half of all human cancers have a genetic lesion involving this gene,3 including CNS cancers.4 An abnormal TP53 gene can be inherited through the germline or can develop through a somatic mutation. The gene also can be lost through the loss of chromosome arm 17p or can be inactivated through a structural rearrangement of chromosomes. Each event either results in the loss of the p53 protein or creates a mutated form that may function abnormally or not at all.
Tumors without a TP53 mutation can be rendered nonfunctional by a variety of mechanisms.3 Normal or wild-type p53 protein can be inactivated by forming a protein complex with viral oncoproteins or with another cellular gene product. One example is the regulation of the p53 protein by the mouse deleted mouse 2 (MDM2) protein. MDM2 binds to wild-type p53.5 This binding inactivates the wild-type p53 so that it cannot interact with other proteins. MDM2 can be amplified or overexpressed in a tumor cell. In primary brain tumors this gene is found to be amplified in some 10% of the cases.6 Aberrant changes involving the p53 protein are considered early changes in the evolution of CNS tumors7 and are associated with a poor prognosis.8
The cell cycle and apoptosis pathways involve many genes and signaling pathways in addition to the ones discussed above. Related literature, however, is sparse, and more investigations are needed before their interactive nature is confirmed. Defining the genetic lesions within tumor types remains an intense area of investigation. The microarray technology with which thousands of genes can be studied at the same time will eventually define the spectrum of genetic abnormalities associated with a particular cancer. The probability of eradicating tumor cells in the future is based on understanding what genes are involved in each tumor type and how the protein encoded by these genes functions in a cell. The identity of a specific nonrandom genetic change in a tumor system has the potential to become a diagnostic and prognostic marker and to serve as a target for the design of new therapies.
 Inherited Intramedullary Spinal Cord Tumors
Inherited Intramedullary Spinal Cord Tumors
The spine, like the brain, is composed of a mixture of cells capable of giving rise to a full spectrum of primary spinal cord tumors. Some spinal cord tumors are associated with genetic syndromes such as neurofibromatosis and von Hippel-Lindau (VHL) disease, but most cases are thought to develop sporadically.
Hereditary cancer syndromes can teach us about the molecular pathways that are central to the life of a tumor cell. The genes responsible for these syndromes often encode proteins that act at critical points in the pathways, controlling fundamental processes such as those discussed earlier; therefore, supporting factors must be established continually to deal with uncontrolled cellular proliferation, differentiation, dedifferentiation, and death to maintain the viability of the tumor mass.
Neurofibromatosis
Neurofibromatosis (NF) occurs as a mutation in either the NF1 or NF2 genes, and each gene plays a different role in the formation of tumors. NF1 is inherited as autosomal dominant and is characterized by multiple neurofibromas, malignant peripheral nerve sheath tumors, optic nerve gliomas, astrocytomas, and multiple café-au-lait spots with a prevalence of 1 in 3000.9 The NF1 gene is mapped to chromosome arm 17q12.10,11 It encodes the protein neurofibromin guanosine triphosphatase, which is an activating protein that affects cellular proliferation and differentiation. Mutations in the NF1 gene do not appear to cluster in hot spots. Of the 300 mutations reported, most have appeared only once.
NF2 is mapped to chromosome arm 22q12.12 It also segregates as autosomal dominant but is much less prevalent, with an incidence of 1 in 40,000.13 The neoplastic and dysplastic lesions of Schwann, meningeal, and glial cells all characterize this disorder. The gene product of NF2 encodes the protein merlin, which is a member of the ezrine-radixin-moesin protein family that links the cytoskeleton to the plasma membrane.
Only a few reports have detailed the genetics of intramedullary spinal cord tumors in relation to NF genes. The scope of studies relating to the spinal cord has been limited, but they have determined that NF1 is predominantly associated with astrocytomas, whereas NF2 is primarily associated with ependymomas. Schwannomas and meningiomas do occur but less frequently than ependymomas.14,15
Like most diseases, phenotypic variability is observed within NF patients. NF, its severity, and its clinical manifestation have been linked to specific point mutations,14–17 splice mutations,18 and missense mutations.17 However, many genetic lesions can be modified by additional genes to produce a clinical phenotype. Modifying genes, genomic imprinting, mosaicism, and environmental influences all contribute and influence outcome. Thus, the analysis of mutations for any one gene may not be entirely predictive of clinical phenotype.
Von Hippel-Lindau Disease
Von Hippel-Lindau disease is an autosomal-dominant disorder whose gene product regulates angiogenesis. The disorder is characterized by the development of hemangioblastomas of the CNS and retina, clear cell renal carcinomas, pheochromocytomas, and pancreatic and inner ear tumors. The VHL protein is a tumor-suppressor gene mapped to chromosome arm 3p25–26.19 The protein encoded in this gene binds to two transcription factors, hypoxia-inducible factor ((HIF)-1a and -2α) and targets them for degradation. HIF-1a and HIF-2α are key proteins that regulate the production of hypoxia-inducible factors such as vascular endothelial growth factor (VEGF), a factor that stimulates blood vessel formation; therefore, mutation, methylation (a process that silences a gene), or loss of the VHL gene by deletion means that the HIF-1α and HIF-2α proteins are not degraded. A continuous signal inducing VEGF is established and results in the formation of excessive blood vessels. Thus, patients with VHL develop hemangioblastomas in the cervical or lumbar spine ~20% of the time. Other sites include the retina, cerebellum, and adrenal glands.20–22
 Sporadic Intramedullary Spinal Cord Tumors
Sporadic Intramedullary Spinal Cord Tumors
Ependymomas
Sporadic intramedullary ependymomas are slow-growing neuroepithelial tumors that account for 50 to 60% of the gliomas in the spine.23 In adults, spinal cord ependymomas occur with about the same frequency as intracranial tumors. In contrast, intracranial tumors in the pediatric population are far more prevalent than spinal cord tumors.
The number of ependymomas analyzed by cytogenetic and molecular analyses is limited, and most investigations included intracranial tumors rather than intramedullary spinal cord tumors. Despite the lack of studies, the data suggest that the most common cytogenetic abnormality in spinal cord ependymal tumors is the loss or rearrangement of chromosome 22.23–27 Less frequent than abnormalities of chromosome 22 were chromosome gains, losses, or rearrangements in chromosome arm 9q and chromosomes 6, 10, 11, 13, and 17.25,28–31 Marker chromosomes (chromosomes that cannot be identified by standard banding techniques) and double minutes (dmins) were observed in only a single case.25
Although their number is still limited, molecular studies performed on spinal cord samples have been more informative than intracranial studies in linking specific abnormalities with a DNA lesion. Ependymomas exhibit specific aberrations that appear to be distinct from those identified in astrocytic gliomas. Like the cytogenetic data, the most common loss of heterozygosity (LOH, loss of genetic material) occurs on chromosome arm 22q.32,33 This finding was not entirely unexpected because of the high incidence of ependymomas in patients with NF type 2. The NF2 gene is mapped to chromosome arm 22q and was considered the likely candidate gene for a mutation in this population of patients; however, this NF2 mutation also appears to be present in sporadic intramedullary spinal cord tumors.34,35
At first this observation was controversial because some studies were unable to confirm these findings.36,37 More recently this problem appears to have been solved. Ebert and coworkers33 studied 62 ependymomas, of which 14 were spinal cord ependymomas. They identified NF2 mutations in 6 of the 14 World Health Organization (WHO) grade II spinal cord ependymomas but in none of 15 intracranial tumors. This investigation supports the hypothesis that NF2 appears to be limited to spinal cord tumors36 and explains why studies including only primarily intracranial ependymomas failed to observe NF2 mutations.
Ebert and co-workers33 also suggested that there might be two distinct subtypes of intramedullary spinal cord tumors: those that possess an NF2 mutation and those with an LOH on chromosome arm 22q but no mutation of NF2. Adult and pediatric tumors may differ with respect to the findings on chromosome 22. A study of 18 pediatric ependymomas identified neither an LOH on 22q nor a mutation in the NF2 gene in the pediatric tissues.37 This observation seems to be confirmed by other studies that included several pediatric samples.35
As indicated, several chromosomes have an LOH in regions where important genes are known to reside. Using these criteria several molecular investigations have analyzed tumor-suppressor gene TP53 (chromosome arm 17p13.1), PTEN (phosphate-tensin),38 also called MMAC1 (mutated in multiple advanced cancers 1)39 or TEP1 (telomerase-associated protein 1)40 (chromosome arm 10q23.3), and the multiple endocrine neoplastic type 1 (MEN1) on chromosome 11. The TP53 studies included an investigation of 31 tumors in which only a single mutation was identified.41 A second study found similar results: Only a single abnormality was observed in 15 tumors.42 Because TP53 is unlikely to be the gene involved, another tumor-suppressor gene continues to be sought on chromosome arm 17p.
Another area of interest is chromosome arm 11q. In a study of 45 ependymal tumors that included 21 intramedullary spinal cord tumors, LOH was assessed for 11q, and a mutational analysis for the MEN1 gene mapped to this region was performed.35 Two WHO grade II ependymomas contained LOH on 11q but no mutations of MEN1. Of the three cases that had both LOH and a mutation of MEN1, each was a recurrent tumor with a diagnosis of WHO grade III. The authors suggested that the loss of the wild-type MEN1 might be involved in the malignant progression of a subset of ependymomas.
Analysis of other tumor-suppressor genes has included PTEN (chromosome arm 10q)33 and tumor-suppressor genes CDKN2A, CDKN2B (chromosome arm 9p21), CDK4 (chromosome arm 12q14), or CCND1 (chromosome arm 11q13.1). No mutations were detected in any of these genes. This finding suggests that they are not the targeted genes in those chromosome regions and therefore do not play an important role in the evolution of ependymal tumors.
Even in larger medical centers, samples of ependymal tumors are limited. Therefore, most investigations contain only a few samples, or they include a mixture of WHO grade II, WHO grade III, and recurrent tumor samples. This situation complicates the assessment of which aberration(s) might be meaningful for diagnosis and prognosis. Thus, another technique, comparative genomic hybridization (CGH), is gaining importance in elucidating the genetic losses and gains in this tumor type.
Comparative genomic hybridization permits genome-wide screening of alterations in copy number (number of alleles). The technique is based on a modification of in situ hybridization in which test DNA (usually tumor DNA) and normal DNA are differentially labeled with fluorescent dyes and hybridized to normal metaphase spreads.43 After hybridization, the color-ratio changes along the metaphase chromosome reveal any differences in copy number between the test DNA and the normal DNA.
A large study of 44 ependymomas included 14 primary intramedullary tumors.44 Copy number abnormalities were identified in all cases. The most notable gains were chromosome 7 (13 samples), 9q or chromosome 9 (in eight tumors), and the X chromosome (in eight tumors). Additional but less frequent gains were on chromosomes 2 (five samples), 5 (five samples), 12 (eight samples), 15q (six samples), and 18 (three samples). The most notable loss was 22q (11 samples), although this report failed to indicate any of the patients were diagnosed with NF. Of these 14 samples, 22q was the only loss identified in seven. The next most frequent loss was 13q and chromosome 13 (four samples). Hirose and coworkers44 also looked for associations among the losses and gains and determined that the gain of 12q was significantly associated with gains on chromosomes 15 and X and the loss on 22q. A second study involving three WHO grade II ependymomas also detected a gain on 12q24 (in all three samples) and a gain on 2p23 in one sample.45 The tumor with the 2q23 amplification recurred and became disseminated throughout the neuraxis. Each sample carried the same 2p23 amplification; however, this study detected no 22q loss in these three tumors.
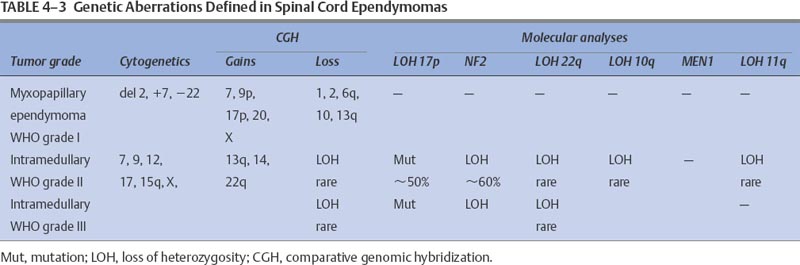
In summary, these data suggest that as in intracranial astrocytomas, there are subsets of ependymomas (Table 4-3). The clinical phenotypes and tumor behavior lend some support to this hypothesis. Evidence also indicates that intramedullary spinal ependymomas differ from intracranial ependymomas with respect to their genetics, age at diagnosis, and prognosis. On average, patients with an intramedullary spinal ependymoma are ~40 years old.46 These spinal tumors are associated with a significantly better prognosis than their intracranial counterparts. Therefore, it is likely that subsets of ependymomas will have genetically different tumor genotypes, and some of these markers may have prognostic value.
Myxopapillary Ependymomas
Myxopapillary ependymomas are slow-growing tumors that primarily occur in young adults and are almost exclusively located in the terminal region of the spine. These tumors have not been subjected to extensive cytogenetic or molecular analyses. Standard banding techniques have revealed a rearranged chromosome arm 1p in a single case47: a dicentric chromosome and a deletion of chromosome arm 11q.48
Molecular studies also show specific genetic alterations in this tumor type. A study of 11 WHO grade I tumors detected LOH on 22q in two samples and on 11q in one sample, but the potential target genes for mutations in these chromosome regions, NF2 and MEN1, had no detectable mutations.35 Other studies have confirmed the absence of LOH on 22q.33,37 Several chromosome regions implicated in other CNS tumors were also negative for chromosome arm 10q,37 and LOH was rare.35
Comparative genomic hybridization studies have begun to provide additional information about potential nonrandom genetic changes in myxopapillary ependymomas. In a study of seven tumors, the most frequent loss was chromosome arm 13q14–31 (six samples), whereas chromosome arm region 9q showed notable gains in five samples.45 Less frequent changes included the loss of 6q (in four samples) and the gain of chromosome 17. However, no loss was observed on 22q in any of these tumors. A second study analyzed six additional myxopapillary ependymomas and, like the previous study, identified no loss on 22q but did find losses on chromosomes 1 (two samples), 2 (three samples), and 10 (two samples).44 These authors also suggested that intramedullary spinal cord ependymomas and myxopapillary ependymomas are different genetic subgroups despite being considered the same histologic entity. Thus, myxopapillary ependymomas differ from intramedullary ependymomas in that they have no loss on 22q, nor do they incur a gain of 15q and chromosome 12. More studies will be needed to verify these findings. However, data suggest that the losses and gains of these two tumor types do indeed differ, and these differences might account for their different clinical behavior.
Subependymomas
Subependymomas are slow-growing, benign neoplasms that account for ~8% of the ependymal tumors in the spinal cord. The most frequent sites involved are the cervical and cervicothoracic regions.49
Several reports detail rare familial cases,50 and this condition has been described in identical twins.51 However, most cases of this rare tumor are thought to develop sporadically.
Cytogenetic and molecular studies are scant and, as of yet, do not demonstrate consistent nonrandom aberrations. The karyotypes of most primary tumors appear to be normal.31,33,52 However, one study of a recurrent tumor identified nonclonal structural abnormalities on the short arm of chromosome 17.52
 Astrocytic Tumors
Astrocytic Tumors
Low-grade diffuse astrocytomas can occur at any site in the CNS, but the preferential site is the cerebral hemispheres. Approximately 60% of all primary intracranial tumors are of this type. Intramedullary spinal cord tumors are far less common, with an incidence of 25 to 30% and a slight male predominance.53 In the pediatric population, however, the incidence (59%) is highest in younger children54 and decreases with age.55
Most cytogenetic and molecular analyses have been performed on intracranial tumors. Based on the few cases that have been analyzed, some if not all of the genetic aberrations associated with intracranial tumors likely play a role in the development and progression of spinal cord astrocytomas. Of all brain tumors this class of tumors has been studied the most extensively, primarily because of their frequency and the availability of tissue. An important concept that has begun to emerge from these analyses is that the genetic differences detected appear to define at least two different pathways that can produce the most malignant form of this tumor, glioblastomas multiforme (GBMs).7 If the patient population has been carefully selected on the basis of clinical and histopathologic data,56,57 the genetic lesions are different in each of these pathways.58
Type 1 or Secondary Glioblastomas
The type 1 pathway is a progressive, stepwise evolution from a low-grade diffuse astrocytoma, to an anaplastic astrocytoma, to a GBM (Table 4-4). Patients diagnosed with this progressive glioma tend to be young (mean age, 39 years) and to have a history of a less malignant tumor at the time of diagnosis.56
The most common mutation or LOH occurs in the chromosome arm of 17p. The target gene is TP53 (17p13.1). There are several so-called “hot spots” in this gene located in codons 175, 248, and 273,59 a highly conserved region spanning exons 5, 7, and 8. Approximately 65% of patients between the ages of 18 years and the mid-40s have a mutation in the TP53 gene compared with older patients with a mean age of 60 years (9%).60,61 A similar observation has been made with immunohistochemical analysis of the p53 protein. These investigations have shown that ~75% of these gliomas have an abnormal accumulation of p53 protein.
TP53 is one of the key genes in malignant transformation, and this event occurs early in the evolution of the type 1 glioma. The p53 protein is a key factor in the DNA repair of damaged cells. The p53 protein inhibits damaged cells from passing beyond the G1 phase of the cell cycle. If the cells are incapable of repair, the p53 protein can induce these cells to die via the apoptotic pathway. Thus, aberrations in this gene are thought to be important in the genesis of low-grade astrocytomas by abrogating apoptosis or by increasing genomic instability.
A second family of genes appears to be important in the evolution of astrocytomas. The platelet-derived growth factor (PDGF) family consists of an A and B chain (PDGFA and PDGFB) and has two receptors, PDGF receptor-α and -β (PDGFR-α and PDGFR-β).62 The PDGF A and B chains dimerize to form AA, BB, or AB homo- or heterodimers. The receptor PDGFR-β binds with AA and AB, whereas PDGFR-α binds with only the BB homodimer. In astrocytomas, the A chain and PDGFR-α are predominantly overexpressed.63 This observation is interesting because the most common chromosomal abnormality identified in astrocytomas involves aneuploidy of chromosome 7,64 the chromosomal location of the PDGF A chain. Studies using fluorescent in situ hybridization (FISH) on low-grade astrocytomas have also demonstrated that the gain (ploidy) of chromosome 7 correlates with shorter survival in astrocytomas.65 This observation may also have relevance for therapy-related issues. In vitro and in vivo treatment with the chemotherapeutic agent BCNU appears to select for a minor subpopulation of cells containing amplified PDGF A and B chains.66 Changes in the expression of growth factors and their receptors also may initiate local environmental changes that begin to stimulate angiogenesis.
Other genetic changes suggest that there may be additional subsets of tumors. In a small number of diffuse astrocytomas, LOH has been identified on chromosome arm 22q. The likely candidate gene for this genetic loss has been the NF2 gene. Extensive analysis of this gene in all grades of gliomas has failed to detect a consistent abnormality.36,67 The LOH reported for chromosome arm 22q occurs more telomeric to the NF2 gene. Thus, a candidate gene or genes to explain this observation await further genetic analysis of these low-grade tumors. Other chromosomes exhibiting LOH in diffuse astrocytomas include chromosome arm 1p, chromosome arm 3p, and chromosome arm 13q. Each LOH identified in these studies represents a probable site for tumor-suppressor genes, but their presence awaits confirmation.68
Anaplastic Astrocytomas
Anaplastic astrocytomas are found in both young and old patients, with the peak incidence occurring in the mid-50s. The frequency of mutation of TP53, the over-expression of the PDGF A chain and PDGFR-α, and LOH on 22q are all similar to those associated with low-grade diffuse astrocytomas. These similarities support the progressive nature of diffuse astrocytomas to a more malignant stage defined as anaplastic astrocytomas.69
Changes that mark the transition from diffuse astrocytoma to anaplastic astrocytoma include LOH on chromosome arms 9p, 11p, and 19q.68 Many of the genetic changes in anaplastic astrocytomas are associated with critical steps in the cell cycle. A key gene in this scenario is the retinoblastoma gene (Rb1) on chromosome arm 13q14, which appears to be altered markedly. The Rb1 protein inhibits the transition of the G0/G1 phase to the S phase. When phosphorylated by any of the CDKs, the Rb1 protein is inactivated (Table 4-4). In-activation of Rb1 allows E2F to promote the cell to progress from G1 to S in the cell cycle. “Upstream” genes such as the INK and Cip/Kip family of genes (Fig. 4-1) can alter the behavior of the Rb1 gene. Thus, the cascade of events leading to the inhibition of the Rb1 protein is controlled by many genes, and the failure of any one gene to function normally permits the cell cycle to continue unchecked. In fact, evidence suggests that it is rare to find more than one DNA lesion in the critical steps of the cell-cycle pathway.70
Deregulation of the cell cycle can be related to LOH or to the amplification of genetic material. An important site of LOH leading to anaplastic astrocytomas occurs on chromosome arm 19q13.2–13.3. The putative tumor-suppressor gene has not yet been identified. This gene appears to be unique to glial tumors and is primarily associated with type 1 gliomas (54%) compared with type 2 gliomas (6%).71
A frequent amplification of a chromosome region occurs on chromosome 12 (12q13–14). This region is amplified in 15% of WHO grade III (anaplastic astrocytomas) and grade IV GBMs.72 The genes MDM2 and CDK4 are mapped to this chromosomal region. Thus, the overrepresentation of whole chromosomes (aneuploidy) or structural rearrangements resulting in the duplication of specific regions of a chromosome can contribute to the amplification of protein(s) affecting the cell cycle.
Glioblastomas Multiforme
Glioblastomas multiforme are considered the most malignant of the astrocytic tumors. The genetic instability of GBMs causes numerous and varied genetic changes, which are reflected in the disparate number of cell types and chromosomal and molecular aberrations (Table 4-4).
Chromosome numbers can range from hypodiploid to several hundred chromosomes per cell. Structural rearrangements are also common and highly variable. These structural rearrangements are often complex and involve multiple chromosomes.
The frequency of TP53 gene mutations, LOH, or both is about the same in GBMs as in anaplastic astrocytomas and diffuse astrocytomas; however, LOH for 10p, 10q23.3, and 10q25.3–26 increases substantially,57,60,73 as does the loss of the entire copy of chromosome 10.68 The tumor-suppressor gene PTEN38 was identified on chromosome arm 10q23.3.39 It encodes a dual specificity phosphatase involved in the regulation of cell growth, apoptosis, cell migration, and interactions with the extracellular matrix. It was initially thought that the LOH of PTEN was a late event that initiated an anaplastic astrocytoma to progress to a GBM. When GBMs were selected on the basis of their being type 1 or 2, only a small number of the progressive type 1 gliomas had a mutation in this gene. Most of the LOH was identified in the type 2 GBMs.74 LOH detected in type 1 GBMs was primarily confined to 10q.
| Astrocytoma→ | Anaplastic astrocytoma→ | Glioblastoma multiforme |
| +7 | +7, 10, +20 | LOH10q |
| LOH 17p | LOH 19q | PTEN mutations |
| TP53 mutation | LOH11p | LOH10p |
| LOH 13q | RB1 changes | DMBT1 mutation |
| PDGFA overexpression | CDK4 | DCC Loss of expression |
| PDGFR-α overexpression | LOH 10q | PDGF-α amplification |
| LOH1p | LOH13q | |
| LOH3p | LOH22q | |
| 12q13 amplification |
A second gene, DMBT1 (deleted in malignant brain tumors 1), may be another important change on chromosome arm 10q in type 1 gliomas. Aberrations in this gene are thought to contribute to genetic instability. DMBT1 is located at 10q25.3–26 and is considered the candidate tumor-suppressor gene found deleted in ~38% of type 1 GBMs.75,76
Platelet-derived growth factor and its receptors have few mutations; however, most GBMs overexpress at least one PDGF chain and its respective receptor. The most common form of overexpression is identified with the PDGF A chain and PDGFR-a growth factor receptor.77,78 The PDGFR-a is capable of binding all three isoforms, and this ability suggested the formation of an autocrine loop for this growth factor similar to the autocrine behavior of epidermal growth factor receptor (EGFR) discussed later. Immunohistochemistry and in situ hybridization further clarified that the PDGF A chain and the PDGFR-α were preferentially expressed in tumor cells where the overexpression of the PDGF B chain and the PDGFR-α is primarily associated with proliferating endothelial cells within the tumor.79–81 Normal brain also expresses the PDGFR-a and the PDGF A chain; however, the PDGFR-a binds only with the PDGF B chain; therefore, it is not active in normal brain.79 This finding suggests that the preferential expression of the PDGFR-α in the endothelial component of gliomas is related to angiogenesis in these high-grade tumors.
Another aberrant gene in GBMs that might contribute to their malignant and invasive phenotype is DCC (deleted in colon cancer), mapped to chromosome arm 18q21. This is a transmembrane cell adhesion molecule of the neural cell adhesion molecule (NCAM) family. As the name implies, DCC was discovered in colon cancer. High-grade gliomas have abnormalities of chromosome 18, and several molecular studies have indicated that it is deleted in ~50% of GBMs82–84 and in some grade II tumors.84 This finding has led some investigators to speculate that cell guidance molecules can be involved in tumorigenesis.
Loss of heterozygosity in chromosome 19, which carries a putative tumor-suppressor gene, is similar to that found in anaplastic astrocytomas when tissue representing type 1 or 2 GBMs is considered. Because most investigations preceded the suggestion that there are subsets of gliomas, the reported frequency is lower in GBMs than in anaplastic astrocytomas. This issue requires further analysis.
Type 2 or Primary Glioblastomas
The second pathway that produces GBMs appears to arise de novo or very rapidly from a preexisting tumor (Table 4-5). Histopathologically, this tumor is indistinguishable from type 1 glioblastomas even though no evolutionary component is apparent. Type 2 tumors are usually found in older patients (mean age, 55 years) who lack a history of a lower-grade tumor.85
| Normal astrocytes → Glioblastoma multiforme |
| +7 EGFR amplification; overexpression and truncation MDM2 amplification overexpression -9p CDKN2A/p16 deletion LOH 13q RB1 mutation LOH 10p and 10q -10 PTEN mutation PDGFA overexpression |
In contrast to type 1 GBMs, the most common genetic aberration in primary type 2 GBMs is the amplification of EGFR in ~40% of the tumors (Table 4-5). More than half of these tumors also have a rearrangement of this gene.86 This mutated form of the EGFR is associated with a high level of tyrosine kinase activity in the absence of the EGF ligand, which essentially keeps this receptor in a “turned on” autocrine mode. Potentially, the amplification of EGFR can override the normal negative regulation of the PTEN gene product.87 Epidermal growth factor receptor is mapped to chromosome arm 7p13-p11. The DNA copy number may be normal, but the gene also can be overexpressed, a feature identified in more than 60% of these tumors.69
Loss of heterozygosity on two regions of chromosome 10, 10p and 10q, is almost entirely restricted to primary type 2 gliomas.74 It has been suggested that the loss of whole chromosome 10 is a major factor in the evolution of highly malignant GBMs. The loss of chromosome 10 is not a feature associated with diffuse astrocytomas. Thus, the loss of chromosome 10 may permit the abrupt change from a low-grade to a high-grade mass. Unlike secondary GBMs, primary type 2 GBMs tend to demonstrate the loss of the entire chromosome 10 rather than the loss of 10q associated with type 1 GBMs.88
The MDM2 gene product also appears to be restricted to the primary de novo pathway.5 The amplification of the MDM2 gene by binding to p53 essentially inactivates this protein. This mechanism serves as an alternative that allows a tumor cell to be removed from the control of normal TP53 gene expression. In fact, tumors that over-express MDM2 usually lack a mutation or LOH in TP53.5
Genes involved in the normal function of the cell cycle also appear to be a feature of primary type 2 GBMs. These genes include the CDKN2 locus that codes for p16INK4A and p14ARF, CDK amplification, and LOH and mutation of Rb1 (Fig. 4-1). When one selects samples on the basis of clinical criteria, these genes are primarily restricted to type 2 or primary GBMs.69
Platelet-derived growth factor A (7q11–13) is also overexpressed in type 2 GBMs, as are many other growth factors.89 Its autocrine regulation suggests that like EGFR, it could provide a selective growth advantage to tumor cells.
 Mesenchymal Tumors
Mesenchymal Tumors
Meningiomas
The incidence of meningiomas is ~15% of all primary brain tumors.90 Spinal cord meningiomas occur less frequently than intracranial tumors, but they constitute the largest group of tumors in this region. They can occur in children and adults. The incidence in children is less than 2%, but they would tend to be the more aggressive forms. In adults the peak incidence is in middle age. The incidence in females is higher than in males (2:1 ratio), especially with spinal cord meningiomas, which are generally localized to the thoracic region. Meningiomas can be familial and are largely associated with von Recklinghausen’s disease. Most cases, however, are sporadic.
The current grading system recognizes three grades of meningiomas: benign (I), atypical (II), and anaplastic (III). Their histologic features were described in the previous chapter. About 94% of the tumors are benign, 5% are atypical, and 1% are anaplastic.90,91 The atypical and anaplastic meningiomas pose the greatest risk of recurrence. Prognostic markers indicating those at risk for these more aggressive forms would be helpful in ensuring appropriate treatment for patients, especially children. Meningiomas have a wide range of histopathologic appearances. The most common types are meningothelial, fibrous, and transitional subtypes.91
Meningiomas are relatively slow-growing tumors. Cellular proliferation increases from benign tumors to atypical to anaplastic tumors. Meningiomas were one of the first solid tumors in which a nonrandom loss of a whole chromosome was identified92 (Table 4-6). Along with the deletion of chromosome 22, monosomy 22 is the most consistent chromosomal aberration.93 Nonetheless, this abnormality is independent of the tumor’s biologic behavior because progression is thought to be more closely associated with chromosomal rearrangement, loss of chromosome arms 1p and 14q, or both.94–96 The cytogenetics of meningiomas are reviewed extensively elsewhere.97
In more than half of the meningiomas analyzed, molecular studies have detected genetic abnormalities on chromosome arm 22q12.98–102 Additional molecular changes are reflected in atypical meningiomas. The most common LOH is identified in 1p, 6q, 9q, 10q, 14q, 17p, and 18q.100,103–106 Anaplastic meningiomas exhibit LOH for these same chromosome regions in addition to the gain of genetic material for chromosome arm regions 20q, 12q, 15q, 1q, 9q, and 17q.107
Approximately 60% of sporadic meningiomas have LOH on chromosome arm 22q and mutations in the NF2 gene.98,99,102,108 Within the different types of meningiomas, the frequency of NF2 mutations varies. Seventy to 80% of fibroblastic and transitional meningiomas carry NF2 gene mutations, in contrast to 10 to 20% of NF2 mutations in meningothelial meningiomas.109 The position of NF2 mutations differs, but each genetic change produces a nonfunctional truncated protein. Analysis of tumor samples has also shown that the normal allele is missing. Such cells have completely lost the merlin protein, supporting the hypothesis that this loss is the major mechanism leading to the formation of meningiomas.110
| Definition | Cytogenetic aberrations* | Molecular genetic aberrations |
| Meningioma WHO, grade I | ~50% −22 | LOH 22q12, NF2 mutation |
| LOH 18p11.3, target gene DAL−1 | ||
| Atypical meningioma | −14, −18 (rare), ~50% −22 del(1p) | Loss 1p, 6q, 10q, 14q, 17p, 18q |
| WHO, grade II | Gains 1q, 9q, 15q, 17q, 20q | |
| Gain 12q associated with amplification of CDK4 and MDM2 | ||
| Anaplastic meningioma | 10 | Loss 6q, 9p, and 14q |
| WHO, grade III | Amplification 17q23 | |
| TP53, PTEN mutations (rare) | ||
| CDKN2A mutations (rare) | ||
| Aberrant expression of growth factors, PDGF, IGF |
*Whole chromosome loss.
LOH, loss of heterozygosity.
Like ependymomas, meningiomas are neoplasms associated with NF2. The target gene is NF2, which encodes the protein merlin.108 While NF2 is the target gene in 60 to 70% of cases, other mechanism(s) must explain the remaining 40% of sporadic cases and families with multiple meningiomas (familial) that do not have a germline mutation in the NF2 gene.111 One explanation may be attributed to the family of membrane proteins to which NF2 belongs. The NF2 gene product, merlin, is a member of the protein 4.1 family and shares the greatest sequence homology with ezrin, radixin, and moesin.12 Another novel member of this protein family, DAL-1 (differentially expressed in adenocarcinoma of the lung), mapped to chromosome arm 18p11.3, was lost in ~60% of sporadic meningiomas.112 Like merlin, this protein is responsible for linking the actin cytoskeleton to cell-surface glycoproteins. Because the loss of expression was detected in WHO grade I meningiomas, these investigators suggest that, like NF2 mutations, this is an early event in the development of meningiomas. They concluded that both merlin and DAL-1 are critical growth regulators in the pathogenesis of meningiomas.
Another possible explanation is the presence of additional tumor-suppressor genes residing on chromosome 22. Rey and coworkers113 identified LOH between 22q12.3 and the telomere, a region that does not involve the NF2 gene. Several candidate genes mapped to this region, including the (3-adaptin gene114 and MN1,115 are under consideration.
Although chromosome arm 22q remains a site of intense investigation, molecular studies have also detected LOH on chromosome arm 1p116–119 and 14q.103,105,120 Genes localized to the deleted region on 1p include TP73 mapped to 1p36119 and p18/CDKN2C/INK4 mapped to 1p32118,121 Investigations probing for mutations of these genes suggest that they are not the targeted tumor-suppressor gene involved in meningioma development because only an occasional mutation has been detected.
Loss of heterozygosity has also been reported on chromosome arms 9q, 10q,100,104,105 and 17p.122,123 These genetic aberrations have each been associated with aggressive or invasive meningioma phenotypes, suggesting that one or more of these changes are key to the progressive evolution of these tumors. Of particular interest was the LOH on 17p and the possible role for mutations of the TP53 gene; however, analysis of the TP53 gene detected no mutations in exons 4 to 8, the regions known to contain most genetic lesions.124 Despite the negative findings for TP53 mutations, immunohistochemical staining performed on primary and recurrent meningiomas detected the aberrant expression of the protein p53.124–126 This finding was considered to have prognostic significance for predicting the recurrence of meningiomas.125
One explanation for the aberrant expression of p53 protein lacking TP53 gene mutations is related to the function of the MDM2 gene, a phosphoprotein that can form a complex with both wild-type and mutant p53 protein.127 Under normal conditions wild-type p53 tumor-suppressor gene and the MDM2 oncoprotein form a feedback loop that upregulates the production of cellular MDM2, blocks p53 activity, and promotes p53 decay, thus preventing an accumulation of p53 protein; however, with genotoxic stress such as DNA damage, the interaction between p53 and MDM2 can be disrupted by modification of either protein. Therefore, if both proteins remain phosphorylated because of damage, p53 does not degrade and high levels of p53 coexist with high levels of MDM2, a condition observed with immunohistochemical staining.124
Meningiomas also appear to contain the aberrant expression of several growth factors (Table 4-6). In particular, the B chain of PDGF and the PDGF-β receptor have been identified by several studies.128–131 An immunohistochemical study further supports a possible autocrine loop for PDGF. Fresh meningioma tissue samples express the PDGF B chain and PDGF-β receptor aberrantly, and this expression was localized to tumor tissue rather than to normal stroma.132 Other growth factors identified in meningiomas include insulin-like growth factor I(IGF-I),133–137 insulin-like growth factor II (IGF-II),134 transforming growth factor α (TGF a),138 transforming growth factor β (TGF-β),139,140 VEGF,141,142 and basic fibroblast growth factor (bFGF).142 The role each growth factor plays in the progression of meningiomas awaits further clarification. Most of the investigations detailed above primarily used intracranial meningiomas; therefore, it is not yet possible to determine if spinal cord meningiomas differ from intracranial tumors with respect to these genetic markers.
 Cranial and Peripheral Nerve Tumors
Cranial and Peripheral Nerve Tumors
Schwannomas
Schwannomas are WHO grade I tumors of the peripheral nerves. They account for ~8% of intracranial tumors143 and ~29% of primary spinal tumors.144 Their incidence is highest in patients with NF2. In the absence of NF2, multiple subcutaneous schwannomas are indicative of a recently described, rare genetic disorder called schwannomatosis.145
Schwannomas most often arise from peripheral nerves in the head and neck and from spinal and cranial nerves. The preferred sites are the vestibular division of the eighth cranial nerve and, less frequently, the fifth cranial nerve. Most tumors are sporadic, but bilateral tumors occur and are most closely associated with NF2. The target gene is the tumor-suppressor gene NF2 that encodes the merlin protein (also termed schwannomin).
Cytogenetic and molecular analyses have identified the involvement of chromosome 22, either by its loss146 or as a LOH of 22q.147,148 The target gene for this LOH is the NF2 gene in which a mutation has been identified in ~60% of schwannomas.12,147,149–151 These genetic alterations each produce a nonfunctional protein. Tumors with this genetic alteration also lose the wild-type allele. Consequently, the tumor cell is devoid of merlin protein, a feature that has been confirmed by immunohistochemical investigations.152–154 Thus, a mutation in the NF2 gene appears to be a major mechanism leading to the development of schwannomas. Further investigation of tumors that have lost 22q but lack a detectable NF2 mutation is needed. They suggest that another tumor-suppressor gene(s) capable of inducing tumor formation is yet to be identified on chromosome arm 22q. Aside from chromosome arm 22q, no consistent genetic alteration has been identified except for an occasional loss of 1p.155
Neurofibromas
Neurofibromas are typically associated with von Recklinghausen’s neurofibromatosis type 1 (NF1). NF1 can be caused by a germline mutation or by a new mutation of the NF1 gene mapped to chromosome arm 17q11.2 by linkage analysis156,157 and by the identification of two translocations in NF1 patients.158,159 This autosomal-dominant disorder has multiple manifestations, including mental retardation, learning disabilities, macrocephaly, bone abnormalities, and the presence of multiple tumors affecting various organ systems.160 Different cell types can be involved, but the most common abnormalities occur in cells derived from the neural crest. This disorder is characterized by the presence of café-au-lait spots, auxiliary freckles, iris hamartomas (Lisch nodules), and neurofibromas (mixtures of Schwann cells and fibroblasts). It is a common disorder with an incidence of ~1 in 4000.161
The NF1 gene encodes the protein neurofibromin, a cytoplasmic protein that has several isoforms. It is expressed ubiquitously in most mammalian tissues, although the highest levels of expression are in the CNS and peripheral nervous system. It is a large gene consisting of 60 exons, and mutations do not appear to cluster in any one exon.162,163 The large size of this gene, its high rate of mutation, and the predominance of small deletions/insertions and point mutations complicate the detection of NF1 mutations. Nonetheless, some 300 have been reported.164 The protein neurofibromin is a guanosine triphosphatase (GTPase)-activating protein that inhibits Ras-mediated signal transduction.165–168 Inactivating mutations of the NF1 gene leads to the loss of neurofibromin function so that control over this signaling pathway, which is involved in cellular proliferation, is lost.
Cytogenetic studies of benign neurofibromas have largely been uninformative. Most of the cells reported tend to have normal 46XX or 46XY karyotypes,169 or the reported changes are inconsistent.170–172 In contrast, neurofibromas that evolve to malignant nerve sheath tumors carry both structural and numerical abnormalities.169,171
Molecular analyses have focused on mutations within the NF1 gene. Tumors known to carry germline mutations have lost the wild-type NF1 allele within their neurofibromas.173,174 This observation indicates that this tumor is generated according to the two-hit hypothesis.175–178 Given the mixed cellular composition of neurofibromas, early studies suggested that these tumors might be polyclonal.170 More recent studies clearly indicate a monoclonal origin.179 The only cell that carries the mutation within the mixture of cells is the Schwann cell.180
Although the NF1 gene is implicated in the genesis of neurofibromin, it is unlikely that the loss of the NF1 gene alone promotes progression to a malignant NF1 tumor. Because tumor progression is a multistep process, additional genetic changes are presumed to follow. One gene examined in relation to the progression of neurofibroma is the TP53 gene localized to chromosome 17 (17p13), a gene known to be involved in the process of oncogenesis. Investigations have been limited, but studies have determined that TP53 is aberrant in neurofibrosarcomas,181–183 further supporting the evidence that this gene is involved in the progression of neurofibroma. The only other chromosome implicated in playing a role in tumor progression is chromosome 22. Neurofibrosarcomas carry an LOH on 22q11–q13.1,184,185 suggesting that this region also may be involved.
NF1 variants that do not co-segregate with the NF1 gene locus also occur.186,187 One explanation is somatic mosaicism, which can extend the range of variability observed 183,188,189
Perineuriomas
These exceedingly rare benign tumors are composed entirely of neoplastic perineurial cells that proliferate and form soft tissue tumors unassociated with a nerve. They are WHO grade I and represent less than 1% of nerve sheath tumors. The proliferating cells form concentric layers around nerve fibers and are easily recognized with appropriate immunohistochemical staining because they contain vimentin and epithelial membrane antigen.91
p53 protein and S-100 have also been evaluated.190 Eight clinically and morphologically typical perineuriomas from three males and five females (age range, 11 to 38 years) were studied by histologic, immunohistochemical, and ultrastructural methods. All tumors were intraneural and involved extremities (two sciatic, one median, one femoral, one peroneal, one brachial plexus, one ulnar, and one radial). These tumors were S-100 protein-negative in contrast to the S-100 protein-positive Schwann cell sheaths. MIB-1 antigen labeling ranged from 4 to 17%. Six cases were stained for p53 antigen. Two cases showed no staining, and two others showed only rare staining. The last two cases demonstrated a scattered staining pattern of immunoreactive nuclei.
Because this tumor is rare, only a few reports have detailed cytogenetic abnormalities. In one study an abnormal clone was identified, and the analyzed metaphases were homozygously deficient for the region 22q11.2qter. In a second case, 53% of the interphase nuclei showed three FISH signals with a chromosome 14/22 probe. This finding suggested monosomy either for the centromere of chromosome 14 or for that of chromosome 22 190 In the second study an abnormality of chromosome 22 was identified with FISH using a probe specific for the M-bcr locus, which maps to the chromosome band 22q11. In the two tumors analyzed, a high percentage of nuclei had only one M-bcr signal (44% and 96%).191 Thus, both studies suggest an abnormality involving chromosome 22, although no candidate genes have been proposed.
Malignant Peripheral Nerve Sheath Tumors (MPNSTs)
Malignant tumors that arise from a peripheral nerve or that show nerve sheath differentiation are termed MPNSTs. They correspond to WHO grades III or IV. These tumors represent ~5% of malignant, soft tissue tumors.192 About two thirds of MPNSTs arise from neurofibromas with an NF1 abnormality.193–195 They also can arise de novo from peripheral nerves.91,196
Cytogenetic reports have often demonstrated complex karyotypes with multiple numerical and structural rearrangements.197–199 The more common modal chromosome numbers are hypodiploid (less than 46 chromosomes per cell) or triploid (~69 chromosomes per cell), whereas structural rearrangement involving chromosome loss, rearrangement, and translocation involves all chromosomes. Patients with MPNSTs also diagnosed with NF1 often have structural abnormalities of chromosome 17198 in addition to changes associated with chromosome 22.198,199 No apparent differences were observed in the karyotypes of tumors arising sporadically from those associated with NF1.
In patients with NF1, both NF1 alleles are missing or inactivated. As stated, this gene represents an early change and is instrumental in the development of neurofibromas.200 Many sporadic tumors also have alterations at the NF1 locus, but additional changes are required to transform these tumors into MPNSTs. Malignant progression has now been linked to genetic alterations occurring in cell-cycle genes. Alterations in the TP53 gene and in p53 protein expression are common in MPNSTs.201–203 About 50% of MPNSTs also have a homozygous deletion of the CDKN2A gene, which encodes the p16 cell-cycle inhibitory molecule.204,205 Neither of these gene changes occurs in neurofibromas and so are thought to be associated with the malignant transformation of neurofibromas; however, the extensive cytogenetic changes in these tumors involving numerous other chromosomes suggest that additional genes involved in malignant progression will be identified.
 Peripheral Neuroblastic Tumors
Peripheral Neuroblastic Tumors
Neuroblastic tumors outside the CNS are clinically important neoplasms. Olfactory neuroblastomas are rare tumors. In contrast, neuroblastic tumors of the sympathetic nervous system, and the adrenal gland are considered the most common pediatric tumor of the nervous system. This group of embryonal tumors displays a broad range of neuronal differentiation. Histology and grading define three major types of neuroblastic tumors: neuroblastomas, ganglioneuroblastomas, and ganglioneuromas. Considerable research has been performed on neuroblastomas, but little is known about the genetic aberrations associated with either ganglioneuroblastomas or ganglioneuromas.
Neuroblastomas
Neuroblastomas are embryonal tumors derived from the neural crest. The primary sites include the adrenal glands and sympathetic nervous system, with less frequent involvement of the abdominal, thoracic, cervical, and pelvic sympathetic ganglia.91 Considerable variability is associated with this tumor. Children older than 1 year often manifest with disseminated metastatic disease that tends to be more aggressive than that found in children younger than 1 year. The latter present with a lower stage of the disease, benefit from therapy, and are often cured. In fact, many tumors regress spontaneously. Consistent with this clinical heterogeneity, investigations have attempted to identify the genetic alterations underlying these different clinical outcomes.
Most neuroblastomas are thought to be sporadic, but several reports are consistent with a hereditary predisposition.206 Familial patterns of inheritance appear to be associated with a median age at diagnosis of 9 months compared with 22 months in the general population. A genome-wide investigation identified linkage of familial neuroblastoma in multiple families to 16p12–13, suggesting that this region of the genome may harbor a locus for neuroblastoma predisposition.207
No single abnormality has been present in all cases, nor has one been identified as the causative event in initiating the disease. Abnormalities detected through flow cytometry, cytogenetics, and molecular genetics suggest that there are distinct differences in these tumors and that these differences are associated with tumor staging and aggressiveness; however, the genetics of neuroblastomas have been linked to tumor behavior (Table 4-7). The genetic abnormalities that have prognostic importance include ploidy, deletion of 1p, amplification of MYC-neuroblastoma (MYCN), trisomy of 17q, and LOH at 11q.
Numerical abnormalities of chromosomes are well known in tumor biology. In neuroblastomas the ploidy of a tumor indicates how that tumor will behave. Like other tumor systems, neuroblastomas can have a wide range of modal chromosome numbers. Some tumors are in the diploid range (35 to 57 chromosomes per cell), triploid range (58 to 80 chromosomes per cell), or hyperdiploid range (81 to 103 chromosomes per cell).208,209 At about the same time, flow cytometry investigations were performed on a large number of patients.210 Together, data from these cytogenetic and flow cytometry studies suggested that patients with neartriploid tumors had a more favorable outcome than those with near-diploid or near-tetraploid tumors.211
| Favorable indicators | Unfavorable indicators |
| Age less than 1.5 years | Older than 1.5 years |
| Poorly differentiated subtype | Undifferentiated subtype |
| Low mitotic index | High mitotic index |
| No MYCN amplification | MYCN amplification |
| No deletion of 1p | Deletion of 1p |
| Hyperdiploidy | Diploidy |
| Triploidy | Tetraploidy |
| TRKA expression | TRKB expression |
MYCN, MYC neuroblastoma; TRKA, neurotrophic tyrosine kinase receptor type 1; TRKB, neurotropic tyrosine kinase receptor type 2.
In infants the DNA content of tumor cells provides important information that can be used to determine the lesion’s response to a particular chemotherapeutic agent as well as outcome.210,211 However, the DNA index of neuroblastomas loses its prognostic value when children are older than 2 years. This loss is partially attributed to differences in the karyotypes of cells prepared from infants, which primarily contain numerical changes in chromosomes, compared with adults who exhibit both numerical and structural alterations of chromosomes.
Initially, the deletion to chromosome arm 1p was detected by cytogenetic analysis.212 This deletion was often associated with unresectable tumors and metastatic disease, whereas tumors with an intact chromosome tended to be localized and to be associated with a clinically favorable outcome.213 The breakpoints on 1p are quite variable, suggesting that more than one tumor-suppressor gene might lie in this region—one gene located at 1p36 and a second at 1p32.214–216
Considerable debate also arose over whether a 1p deletion alone had prognostic value or only when combined with MYCN amplification. Kaneko and coworkers217 indicated that a 1p loss in diploid tumors is predictive of a poor survival, whereas a 1p loss from tumors that are trisomic for chromosome 1 appears to have no significance in predicting tumor behavior.
A cell line study offered further clarification. Cell lines that lacked MYCN amplification contained only a small deletion spanning 1p36.23–33. In contrast, deletions in cell lines with MYCN amplification involved a larger region—1p35–36.1 to the telomere.218 The parental origin of the deleted 1p region and whether this 1p LOH was subject to genomic imprinting (a condition in which there is a selective loss of one parental type)219 were also unclear. In tumors with normal MYCN expression, the 1p loss was restricted to a maternal loss in 16 of 17 cases analyzed.216 However, if MYCN amplification occurred with a 1p loss, the LOH appeared to be random in the 20 cases analyzed,216 a finding suggested earlier by Cheng and co-workers.218
These observations may also be explained by the presence of two tumor-suppressor loci on chromosome arm 1p. The more distal one at 1p36.2–3 may be subject to genomic imprinting and is not associated with MYCN amplification. The second is located at 1p35–36.1. It appears to have random LOH and is associated with MYCN amplification.214–216,218
The loss of 1p is frequently associated with an unbalanced translocation involving the q arm of chromosome 17, the common rearrangement being a der(1)t(1,17)(p36;q21).220–222 This translocation has been associated with poor clinical outcomes.223,224 Other studies, however, suggest that outcomes appear to be the same whether these two features occur independently or are in the form of the translocation.225 Although 1p loss was considered the most frequent abnormality, newer molecular cytogenetic techniques such as FISH and CGH confirm that aberrations of 17q are the most frequent genetic abnormality, occurring in ~50% of the tumors.223
Typically, MYCN amplification is associated with the selective increase of gene copy number.226 Amplification associated with an increase in the number of genes is also associated with dmins and homogeneously staining regions (hsrs). Biedler and Spengler227 noted these structures in a neuroblastoma cell line and suggested that they were a form of gene amplification. Other investigations showed that these structures contained sequences that were homologous to the human cellular oncogene MYC228 mapped to chromosome arm 2p23–24.229–231 The MYCN amplification that occurred in neuroblastomas was seldom associated with this gene location, but it was associated with the dmins and hsrs.230–232
As additional investigations were performed, it became apparent that MYCN amplification strongly correlated with stage 3 and 4 tumors and was only rarely observed in stage 1 and 2 tumors.233 When identified in a low-grade stage 2 tumor, MYCN amplification was predictive of a poor outcome. Thus, MYCN amplification is one of the most important indicators for a rapidly progressive disease that usually results in a fatal outcome.234–236
In a study of 349 cases, Guo and co-workers237,238 found the loss of 11q in 41% of the tumors. Most of this loss was localized to 11q23.3. The loss of 11q tends to be associated with poor clinical outcomes. Frequently, an 11q loss occurs with a 14q loss (25%) but has an inverse relationship with a 1p loss and MYCN amplification. This relationship suggests that these tumors may represent subsets of neuroblastomas.239–242
Other sites of LOH include 2q,243 3p,244,245 4p,245,246 9p 246,247 14q 239–241,245,248–251 16p 245,252,253 and 18q 254
In addition to the reported LOH there is also the abnormal gene expression of telomerase, TRK, CD44, and multidrug-resistant associated protein (MRP) in a small number of tumors. In normal cells the ends of chromosomes shorten with each cell division. Eventually, this gradual removal of the ends of chromosomes triggers cell senescence or apoptosis. The enzyme telomerase has the ability to rebuild the ends of a chromosome. Thus, the abnormal expression of this gene is one of the factors thought necessary to immortalize a cell. In some neuroblastomas telomerase is overexpressed, and high values in stage 4 disease are associated with fatal outcomes.255,256
TRK is a transmembrane tyrosine kinase that acts as a receptor for neurotrophin nerve growth factor. Data on this receptor are limited but suggest that the high expression of this gene in low stages of the disease is predictive of favorable outcomes.257
The CD44 gene, a cell-surface glycoprotein involved in cell adhesion, is usually assayed immunohistochemically. Several investigations suggest that the lack of CD44 is associated with poor outcomes.258–261 However, this contention remains controversial because other studies suggest that it has little predicative value.262,263
The overexpression of the MRP gene is associated with cellular resistance to chemotherapeutic agents. It is frequently overexpressed with MYCN amplification264 and is considered to have prognostic significance independent of MYCN amplification.
REFERENCES
1. Sherr CJ. Mammalian G1 cyclins. Cell 1993;73:1059–1065
2. Sherr CJ. The Pezcoller lecture: cancer cell cycles revisited. Cancer Res 2000;60:3689–3695
4. Louis DN. The p53 gene and protein in human brain tumors. J Neuropathol Exp Neurol 1994;53:11–21
13. Gutmann DH. Molecular insights into neurofibromatosis 2. Neurobiol Dis 1997;3:247–261
18. Kluwe L, MacCollin M, Tatagiba M, et al. Phenotypic variability associated with 14 splice-site mutations in the NF2 gene. Am J Med Genet 1998;77:228–233
24. Hamilton RL, Pollack IF. The molecular biology of ependymomas. Brain Pathol 1997;7:807–822
61. Watanabe K, Sato K, Biernat W, et al. Incidence and timing of p53 mutations during astrocytoma progression in patients with multiple biopsies. Clin Cancer Res 1997;3:523–530
101. Meese E, Blin N, Zang KD. Loss of heterozygosity and the origin of meningioma. Hum Genet 1987;77:349–351
138. Halper J, Jung C, Perry A, Suliman H, Hill MP, Scheithauer B. Expression of TGFα in meningiomas. J Neurooncol 1999;45: 127–134
178. John AM, Ruggieri M, Ferner R, Upadhyaya M. A search for evidence of somatic mutations in the NF1 gene. J Med Genet 2000;37:44–49
192. Lewis JJ, Brennan MF. Soft tissue sarcomas. Curr Probl Surg 1996;33:817–872
217. Kaneko Y, Kobayashi H, Maseki N, Nakagawara A, Sakurai M. Disomy 1 with terminal 1p deletion is frequent in mass-screening-negative late-presenting neuroblastomas in young children, but not in mass-screening-positive neuroblastomas in infants. Int J Cancer 1999;80:54–59
226. Schwab M. Amplification of oncogenes in human cancer cells. Bioessays 1998;20:473–479
245. Takita J, Hayashi Y, Kohno T, et al. Allelotype of neuroblastoma. Oncogene 1995;11:1829–1834
256. Poremba C, Willenbring H, Hero B, et al. Telomerase activity distinguishes between neuroblastomas with good and poor prognosis. Ann Oncol 1999;10:715–721
Related posts:
 Lumbosacral and Pelvic Reconstruction
Lumbosacral and Pelvic Reconstruction
 Anatomy of the Spine and Spinal Cord
Anatomy of the Spine and Spinal Cord
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


