Figure 26.1. Hemorrhagic stroke with cytotoxic edema as indicated by hypodensity surrounding the hemorrhage.
While clinical changes are the most important endpoint of cerebral edema, they indicate an advanced stage and therefore should be coupled with other methods such as radiography. Cerebral edema can be identified by computed tomography (CT) observing low density attenuation around the primary CNS insult (Figure 26.1) causing mass effect. Examining its location and dimension, one can estimate the time of the lesion progression and its effects on the surrounding tissue. The effects of the edema can be identified by the amount of sulcal and cisternal effacement or shifting of tissue out of its normal location (Figure 26.2) such as midline shift or temporal lobe herniation through the tentorum cerebelli. MRI can provide further detail of the location and extent of edema with T2 or FLAIR imaging.
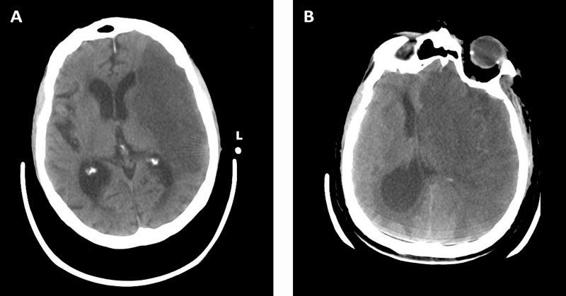
Figure 26.2. Cerebral infarction with edema causing effacement of sulci and ventricles as well as herniation and midline shift of the left hemisphere towards the right.
Cerebral edema can also be measured indirectly by quantifying changes in ICP measured invasively. While the primary insult will have a certain degree of effect in ICP, subsequent changes are more likely due to changes in edema. The intraventricular catheter (ventriculostomy) is a soft tube placed through a burr hole into the lateral ventricle. This not only allows measurement of ICP, but also allows removal of CSF which can relieve pressure caused by edema. The implantable transducer is drilled through the skull with a probe that extends into the parenchyma allowing it to measure ICP but not allowing the removal of fluid. ICP measurement is often the method of choice in diffuse injury such as TBI or global hyponatremia/hyposmolarity caused by metabolic derangements or liver failure, as the CT scan may not show any focal changes. These changes can be followed radiographically in order to determine if an intervention is needed, as well as the response to it.
26.4 Treatment
Now that the causes and methods to measure edema have been explored, we will discuss the treatment of cerebral edema. Several strategies have been developed to treat cerebral edema, both medically and surgically. We’ll focus on medical therapies to remove the excess of fluid related to edema. Surgery can effectively treat edema not only by removing the primary insult, but also by removing a large bone flap allowing decompression of edematous tissue. Alternatively, metabolic suppression through induction of hypothermia or drug induced coma can reduce metabolic activity providing protection from edema.
Osmotic therapy has long been the mainstay of medical therapy of cerebral edema. The use of osmotic therapy is based on water diffusion; two solutions separated by a semi-permeable membrane will allow the flow of solvent from the compartment with lower solute concentration to the one with the higher concentration. In addition to the barrier created by the vascular endothelium, the BBB will prevent solute from following solvent. The same principle that leads to edema can guide its treatment. Accumulation of osmotically active particles from cell death or leakage caused by membrane breakdown creates an abnormal osmotic gradient which is treated with mannitol to create a new one.
Mannitol is the most commonly used osmotic agent for the treatment of cerebral edema. It is a sugar alcohol with the chemical formula C6H8(OH)6. Other examples of sugar alcohols include xylitol and sorbitol. Its properties were first described by Weed and McKibben in 1919 but its use to treat cerebral edema wasn’t common until the 1960s. Its role as the ideal osmotic agent is based on the fact that it has a high reflection coefficient, a measurement of how much a substance is excluded by a membrane. The higher the coefficient, the more likely it is to be excluded by the BBB. Sodium chloride has an approximate coefficient of 0.95 (rated 0 to 1) while mannitol coefficient is 0.90. Because it is largely excluded and is not metabolized by the brain or liver, it is an osmotically active particle as long as the cell membrane and BBB are intact. After administrating a dose, it distributes within a few minutes through the extracellular compartment [10]. The effects of mannitol occur in multiple phases. First, it removes excess fluid by creating an osmotic gradient in favor of the ECF over the ICF. Secondly, mannitol acts as osmotic diuretic: as it is excreted by the kidney, it removes free water and thereby reduces overall volume status. Additional postulated effects of mannitol include increasing RBC rheology, thus reducing blood viscosity. As this leads to increased blood flow, cerebral vascular autoregulation may decrease blood volume, potentially assisting in decreasing ICP. Furthermore, it is postulated that mannitol acts as a free radical scavenger reducing the inflammatory cascade present in cytotoxic edema.
The first question concerning mannitol is, is it effective? The literature clearly shows that it can decrease ICP in TBI [10]. This effect seems to be dose-dependent and higher doses show a more durable ICP control [11]. In respect of cerebral infarction, some animal models show a decrease in peri-infarct edema with mannitol as well as residual deficit, but other models do not show efficacy. The Cochrane review of mannitol in stroke does not find evidence for improvement in outcomes [12]; but the American Heart Association Guidelines still recommends it as a potential therapy based on expert opinion. While osmotic therapy clearly can alter ICP, whether it can cause an improvement in outcome is much harder to demonstrate. In many patients mannitol doesn’t seem to affect a patient’s sodium level: in one series of 167 patients, 73 (44%) didn’t have an increase of sodium greater than 5 mEq/l while 37 patients (22%) didn’t have a rise greater than 1 mEq/l [10].
Typical doses of mannitol are 0.5 to 1.5 g/kg given in four to six hour increments. The main limitations of its use are excessive diuresis causing volume depletion which may lead to renal insufficiency, rebound cerebral edema, and renal failure if it obstructs the renal tubules. Within hours to days of mannitol administration, the brain adapts to the higher osmolality by accumulating yet to be described organic osmoles called “idiogenic osmoles” in an attempt to restore normal cell size. This might explain why its effects seem time and dose limited; however, it is likely that this same phenomenon happens with hypertonic saline or other osmotic therapies.
A possible limitation to mannitol therapy is that as it creates an osmotic gradient most effectively across an intact BBB, it may preferentially extract water from the intact brain and not be as effective in the damaged brain. Consequently, it could theoretically exacerbate the intracranial pressure differential and actually aggravate tissue shifts. This complication may be prevented, however, if mannitol is fully eliminated before administering another dose. The complete clearance of mannitol by the kidneys given sufficient time has been well documented, including MRI spectroscopy showing end stage renal patients treated with mannitol that has leaked into the brain parenchyma who demonstrates absence of mannitol after dialysis [13].
In order to ensure proper clearance and prevention of rebound of cerebral edema, mannitol levels need to be monitored. The goal is to ensure proper clearance between doses. Mannitol cannot be routinely measured in a clinical setting. As a surrogate, serum osmolality is directly measured and compared to the calculated osmolality. The calculated osmolality is simplified to the dominate serum solutes sodium, urea, and glucose. The difference between the measured and calculated osmolality will then be composed of the unmeasured osmoles including mannitol and called the “osmotic gap” (OG).
Osmolality is calculated using the following euation:
2Na+ + (BUN / 2.8) + (glucose / 18)
where BUN = bllod urea nitrogen.
This osmotic gap has been validated by studies as an indicator for mannitol levels with a high correlation of OG to mannitol [14]. Historically, some authors recommended to follow a safety level of osmolarity equal to 320 mOsm/l, but such practice is not well founded as it relies on studies where mannitol was given as a continuous infusion, which is not common in clinical practice. It is important that the osmolality and gap are measured before the mannitol is administered as the gap is used to measure the trough of the drug and not its peak. Gap is not used to measure efficacy but to prevent toxicity by ensuring mannitol is cleared from the blood after each dose. If the current dose is not obtaining the desired effect on the cerebral edema, it can be increased depending on the change of the osmotic gap from its baseline.
The next issue to address is the potential adverse effects caused by inducing a hypernatremic state. To show the extent of the concern, some authors report a mortality of 40% to 60% in those with elevated sodium but this might be do their admitting diagnosis and not the sodium perturberance itself. Hypernatremia can occasionally be found in dehydrated and cognitively impaired patients, but it usually occurs in hospitalized patients. Certainly in a neurologic and neurosurgical ICU hypernatremia is common even in subjects not on osmotic therapies, due to the fact that these patients are obtunded, having central insults causing diabetes insipidus, and central fevers causing insensible fluid loss. Hypernatremia can cause renal failure, gastrointestinal dysfunction, cardiac dysfunction, and worsening delirium. Mannitol itself can be nephrotoxic if it accumulates in the kidneys and shouldn’t be used in people with significant renal impairment. Furthermore, patients with poor renal function caused by such things as hypertension and diabetes require careful monitoring. Typically, in our ICU we use a maxiumum sodium level of 160 mEq/l. In a 6.5 year retrospective analysis of more than four thousand patients, sodium over 160 mEq/l was found to have independent increased mortality on multivariative analysis [15]. Once a patient recovers from their primary insult, it is safe to correct the sodium at a rate no faster than 5-10 mEq/l/day to avoid rebound edema. Additionally, magnesium and phosphorous should be followed on a daily basis to monitor for depletion associated with osmotic dieresis.
Hypertonic saline (HS) has become increasing popular as an alternative to mannitol to treat cerebral edema. Skeptics of HS would point out that mannitol might not be solely effective because of the osmotic effects but because of its other actions as described above, but still consider it a reasonable alternative in patients with renal impairment or refractory to mannitol therapy. It certainly has the advantages of being a volume expander, and therefore might be superior in trauma. In addition, patients with subarachnoid hemorrhage may have edema but also be at risk for developing vasospasm if their volume is depleted by mannitol. Insufficient head to head comparison studies have been done, which are largely limited by varying doses used (how much hypertonic saline is clinically equivalent to how much mannitol?) and the heterogenicity of the patient population treated for cerebral edema. Hypertonic saline does have a slightly higher reflection coefficient than mannitol, and therefore less risk of rebound, but it also has other higher risks. For example, more rapid sodium alterations increase the risk of central pontine myelinolysis. Hypertonic saline leads to significant volume expansion which may lead to heart failure and pulmonary edema if not taken into consideration. The excessive chloride with HS increases the risk of hyperchloremic metabolic acidosis. While it does lack of free radical scavenger properties, it has been correlated with decreased ICP in both adults and children [10]. While mannitol is still the first line drug for cerebral edema, there are patient populations for which HS may be an appropriate alternative.
While mannitol is by far the most commonly used osmotic agent, others have been used. Glycerol is a sugar occasionally used with the theoretical advantage of being metabolized after crossing the BBB making the likelihood of rebound unlikely. It is hard to comment on glycerol efficacy, because of lack of substantial studies or common clinical experience.
Furosemide is commonly used for control of patient’s overall volume status, and its efficacy is certainly well established. Steroids such as dexamethasone are the mainstay of vasogenic cerebral edema as they are well established in stabilizing the integrity of the BBB and thereby reducing fluid influx. Pathologies such as tumor-associated edema are commonly treated with steroids, but they have not proven effective in other pathologies such as stroke. Peritumoral brain edema (PTBE) is related to inflammatory mediators secreted by the tumor such as interleukin [16] and growth factors such as vascular endothelial growth factor (VEGF) causing abnormal vascularization with impaired capillary endothelial tight junctions that leads to vasogenic edema [17]. Steroids have not been shown to be effective in cytotoxic edema probably because the integrity of the BBB is not necessarily compromised in these conditions.
As stated previously, SIADH is a common phenomenon in patients with CNS insults. Hyponatremia can worsen cerebral edema; therefore, it’s worthwhile to discuss its treatment. A relatively new therapy is the V1A/V2 (arginine vasopressor) receptor antagonists such as conivaptan and tolvaptan. They treat SIADH by directly blocking the supraphysiologic ADH at the renal tubule allowing an aquaresis. Previously SIADH was treated with limiting free water intake, hypertonic solutions, and furosemide. Its use requires a careful distinction between cerebral salt wasting and SIADH because in the case of salt wasting syndrome, the further volume depletion with V1A/V2 antagonists would lead to a more severe hypovolemia. Furthermore, its use in subarachnoid hemorrhage patients must be carefully monitored because of the risk of volume depletion contributing to vasospasm. ADH antagonists will cause an aquaresis even in normotremic patients and thereby could be used to treat cerebral edema. Generally speaking, it is better to use ADH antagonists in hyponatremia associated with SIADH, rather than in cerebral edema, but future studies may highlight their role in inducing hypernatremia.
26.5 Conclusions
While cerebral edema seems a relatively simple concept, developments in both clinical medicine and basic science have shown that its causes are as diverse as the disease associated with it and its physiology has complex regulation. We are used to distinguish different types of edema (cytotoxic, vasogenic, hydrostatic, and osmotic); however, most of the diseases encountered in a neurologic ICU show combinations of these types of edema. While many osmotic therapies have been used, mannitol is still the mainstay of treatment, and if used and monitored appropriately it has little risk. The growing use of hypertonic saline will likely continue to increase and in some cases may be a better choice than mannitol. The most exciting development in the science and treatment of cerebral edema is a growing understanding of the role and regulation of aquaporins, whose modulation is already being tested [18] in animal studies and may lead to novel therapies to alter not just the location of fluid by osmotic gradient but augment the brains ability to compensate for edema.
References
1. Strbian D, Durukan A, Pitkonen M, et al. The blood-brain barrier is continuously open for several weeks following transient focal cerebral ischemia. Neuroscience 2008; 153: 175-81
2. Lien YH, Shapiro JI. Hyponatremia: clinical diagnosis and management. Am J Med 2007; 120: 653-8
3. Rama Rao KV, Norenberg MD. Aquaporin-4 in hepatic encephalopathy. Metab Brain Dis 2007; 22: 265-75
4. Frigeri A, Nicchia GP, Svelto M. Aquaporins as targets for drug discovery. Curr Pharm Des 2007; 13: 2421-7
5. Nase G, Helm PJ, Enger R, et al. Water entry into astrocytes during brain edema formation. Glia 2008; 56: 895-902
6. Verkman AS, Binder DK, Bloch O, et al. Three distinct roles of aquaporin-4 in brain function revealed by knockout mice. Biochim Biophys Acta 2006; 1758: 1085-93
7. Romeiro RR, Romano-Silva MA, De Marco L, et al. Can variation in aquaporin 4 gene be associated with different outcomes in traumatic brain edema? Neurosci Lett 2007; 426: 133-4
8. Hirt L, Ternon B, Price M, et al. Protective role of early aquaporin 4 induction against postischemic edema formation. J Cereb Blood Flow Metab 2009; 29: 423-33
9. Chen CH, Xue R, Zhang J, et al. Effect of osmotherapy with hypertonic saline on regional cerebral edema following experimental stroke: a study utilizing magnetic resonance imaging. Neurocrit Care 2007; 7: 92-100
10. Keyrouz SG, Dhar R, Diringer MN. Variation in osmotic response to sustained mannitol administration. Neurocrit Care 2008; 9: 204-9
11. Sorani MD, Morabito D, Rosenthal G, et al. Characterizing the dose-response relationship between mannitol and intracranial pressure in traumatic brain injury patients using a high-frequency physiological data collection system. J Neurotrauma 2008; 25: 291-8
12. Bereczki D, Fekete I, Prado GF, et al. Mannitol for acute stroke. Cochrane Database Syst Rev 2007; 18: CD001153
13. Maioriello AV, Chaljub G, Nauta HJ, et al. Chemical shift imaging of mannitol in acute cerebral ischemia. Case report. J Neurosurg 2002; 97: 687-91
14. Diringer MN, Zazulia AR. Osmotic therapy: fact and fiction. Neurocrit Care 2004; 1: 219-33
15. Aiyagari V, Deibert E, Diringer MN. Hypernatremia in the neurologic intensive care unit: how high is too high? J Crit Care 2006; 21: 163-72
16. Park KJ, Kang SH, Chae YS, et al. Influence of interleukin-6 on the development of peritumoral brain edema in meningiomas. J Neurosurg 2010; 112:73-80
17. Papadopoulos MC, Saadoun S, Binder DK, et al. Molecular mechanisms of brain tumor edema. Neuroscience 2004; 129: 1011-20
18. Okuno K, Taya K, Marmarou CR, et al. The modulation of aquaporin-4 by using PKC-activator (phorbol myristate acetate) and V1a receptor antagonist (SR49059) following middle cerebral artery occlusion/reperfusion in the rat. Acta Neurochir Suppl 2008; 102: 431-6
Related posts:
 Neuroscience Critical Care: Two Experts’ Point of View
Neurocritical Care of Patients With Arteriovenous Malformations
Neuroscience Critical Care: Two Experts’ Point of View
Neurocritical Care of Patients With Arteriovenous Malformations
 Monitoring Cerebral Blood Flow and Cerebral Autoregulation: Basic Principles, Techniques, Common Patterns and Interpretation of Results
Monitoring Cerebral Blood Flow and Cerebral Autoregulation: Basic Principles, Techniques, Common Patterns and Interpretation of Results
 Endocrinology of Acute Brain Injury
Endocrinology of Acute Brain Injury
 Monitoring Brain Chemistry by Microdialysis During Neurointensive Care
Monitoring Brain Chemistry by Microdialysis During Neurointensive Care
 Acute Paraplegias and Quadriplegias of Non-traumatic Cause
Acute Paraplegias and Quadriplegias of Non-traumatic Cause
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


