Chapter 69 Cerebral Palsy
Definition and Clinical Characterization
Cerebral palsy is defined as “a group of disorders of the development of movement and posture, causing activity limitation, that are attributed to non-progressive disturbances that occurred in the developing fetal or infant brain” [Bax et al., 2005]. Virtually all such disturbances occur during or before early infancy. Although tone and postural abnormalities may become more pronounced during early childhood, qualitative evolution is uncommon. The full extent of motor disability may not be evident until the age of 3 or 4 years. Intellectual, sensory, and behavioral difficulties may accompany cerebral palsy, and are especially common in patients with spastic quadriplegia and severe motor disability [Shevell et al., 2009]. Children with cerebral palsy often exhibit mental retardation (52 percent), hearing impairment (12 percent), and speech and language disorders (38 percent) [Ashwal et al., 2004], as well as congenital anomalies [Rankin et al., 2009]. Epilepsy occurs in 34–94 percent of children with cerebral palsy, depending on the study population. Although neurologic impairment beyond motor involvement frequently occurs, the diagnosis of cerebral palsy rests upon the presence of motor disability alone.
Cerebral palsy can be classified by the following attributes [adapted from Bax et al., 2005]:
Cerebral palsy is a clinical syndrome that encompasses a wide range of brain disorders associated with impaired motor function. Thus, the definition of cerebral palsy in research studies often varies, depending on the research question being addressed [Badawi et al., 1998c]. The designation of cerebral palsy as an entity, despite the heterogeneity of underlying etiologic conditions, is valuable because afflicted children commonly have similar medical care, rehabilitation, and social services needs.
Epidemiology
Cerebral palsy occurs in 1.2–3.6 children per 1000 live births. Numerous cerebral palsy registries exist throughout the world [Cans et al., 2004], and population prevalence rates from four continents have remained consistent over several decades. In Denmark, there was a declining birth prevalence between the period of 1983–1986 (3.0 per 1000 live births) and the period 1995–1998 (2.1 per 1000 live births) [Ravn et al., 2009]. In the United States, the prevalence of cerebral palsy increased from 1.7 to 2.0 per 1000 live births between the mid-1970s and late 1980s [Winter et al., 2002], and was estimated to be as high as 3.6 per 1000 8-year-old children in 2002 [Yeargin-Allsopp et al., 2008]. Based on these numbers, approximately 12,000 children with cerebral palsy are born annually in the United States.
Prematurity is the single most important risk factor for cerebral palsy. The risk of cerebral palsy in very low birth weight infants is as high as 4–10 percent, whereas the risk in term infants is only 1.0–1.5 per 1000 live births [Grether et al., 1992; Hagberg et al., 2001; Topp et al., 2001; Wu et al., 2003; Ancel et al., 2006; Moster et al., 2008]. Infants born at 24–26 weeks’ gestation may have as high as a 20 percent chance of developing cerebral palsy [Ancel et al., 2006]. In most epidemiologic studies, preterm infants constitute approximately half of all infants with cerebral palsy. An increase in the prevalence rate of cerebral palsy among preterm infants during the mid-1980s was attributed to increased survival of low birth weight infants [Colver et al., 2000; Pharoah et al., 1990; Topp et al., 2001]. Subsequent longitudinal studies, however, suggest that the rate of cerebral palsy among preterm infants has remained constant or even decreased [Clark and Hankins, 2003; Hagberg et al., 2001; O’Shea et al., 1998; Winter et al., 2002; Platt et al., 2007], though this is not a consistent finding in all populations [Vincer et al., 2006].
Term infants represent more than half of all cases of cerebral palsy [Bax et al., 2006]. The prevalence of cerebral palsy among term infants, 1–1.5 per 1000 live births, has not diminished over the past three decades, despite the introduction of electronic fetal monitoring during the 1970s and the subsequent sharp rise in births by cesarean section [Clark and Hankins, 2003; Colver et al., 2000; Topp et al., 2001; Winter et al., 2002]. Although unchanged prevalence is disappointing in view of the fact that perinatal deaths, stillbirths, and birth asphyxia, as measured by low Apgar scores, all have dropped dramatically in recent decades, other data are more encouraging [Ravn et al., 2009]. Longitudinal data in the United States suggest that a slight increase in cerebral palsy prevalence among infants with normal birth weight was responsible for the significant increase in total cerebral palsy prevalence reported over a 15-year period [Winter et al., 2002]. Although the severity of cerebral palsy within an Australian population increased during this period [Blair, 2001], suggesting that perhaps more infants who would have died in previous times are being saved, workers in the United Kingdom have described a decrease in severity of cerebral palsy among term infants over a similar (18-year) period [Colver et al., 2000].
Male infants exhibit a higher risk of cerebral palsy than female infants [Platt et al., 2007; Wu et al., 2009; Johnston and Hagberg, 2007]. Twins, who constitute 2 percent of the population, also carry a higher risk, and contribute 10–12 percent to the overall prevalence of cerebral palsy [Grether et al., 1992; Bax et al., 2006]. The increased risk of cerebral palsy among infants of multiple gestation is partly due to the higher rate of prematurity, though twins born at term may also exhibit an increased risk for cerebral palsy [Petterson et al., 1993; Pharoah, 2001]. Death in utero of a co-twin, such as from twin–twin transfusion, places the surviving twin at particularly high risk for neurologic and other complications [Grether et al., 1993; Szymonowicz et al., 1986].
Blacks have an increased risk of cerebral palsy when compared with whites [Murphy et al., 1993; Yeargin-Allsopp et al., 2008]. Although this finding is due in part to an increased rate of prematurity, the risk of cerebral palsy among black infants born at term also is elevated [Wu et al., 2003]. Furthermore, several studies suggest that infants born to mothers with lower socioeconomic status exhibit an increased risk of cerebral palsy [Sundrum et al., 2005; Yeargin-Allsopp et al., 2008; Dolk et al., 2001]. In one study that adjusted for potential confounding effects of prematurity, the effect of low socioeconomic status on cerebral palsy risk was only partly accounted for by lower birth weight and gestational age [Sundrum et al., 2005].
Life expectancy of patients with cerebral palsy is related to the type of involvement and the severity of motor disability. Severe quadriplegia has been associated with a shortened life expectancy. Other significant variables include associated disabilities and availability of quality medical care. The risk of death is highest in the first 5 years of life [Blair et al., 2001]. As mortality data have become available, it has become clear that, with reasonable medical attention, a majority of affected persons will survive into adult life [Evans et al., 1990; Eyman et al., 1990; Hutton et al., 1994; Plioplys et al., 1998; Strauss et al., 1998; Plioplys, 2003].
Etiology
A wide range of causative disorders and risk factors have been identified for cerebral palsy. Box 69-1 lists some of the more common etiologic categories and risk factors, broadly classified into the following groups: perinatal brain injury, brain injury related to prematurity, developmental abnormalities, prenatal risk factors, and postnatal brain injury. These factors may coexist and interact with each other in contributing to the pathogenesis of brain injury resulting in cerebral palsy.
Box 69-1 Selected Etiologic Disorders and Risk Factors in Cerebral Palsy*















TORCH, toxoplasmosis, other infections, rubella, cytomegalovirus infection, herpes simplex.
Perinatal Brain Injury
Intrapartum hypoxia-ischemia is a well-described cause of cerebral palsy, especially in the setting of an acute intrapartum event such as uterine rupture, placental abruption, or cord prolapse. The extent to which hypoxic-ischemic brain injury is responsible for cerebral palsy, however, has been a major source of controversy. Epidemiologic studies suggest that in a minority (between 6 and 28 percent) of affected children, cerebral palsy is due to perinatal asphyxia [Hagberg et al., 2001; Nelson and Grether 1998]. The form of cerebral palsy most often associated with hypoxic-ischemic brain injury is spastic quadriparesis.
The term perinatal asphyxia is confusing and deserves further clarification. Traditionally, this term has been defined by clinical signs and symptoms, including low Apgar scores, meconium-stained amniotic fluid, and low umbilical cord pH. Yet the clinical findings used to define this term are not specific for hypoxic-ischemic brain injury [Blair, 1993]. Although various groups of investigators have defined specific criteria that can be used to establish intrapartum hypoxia-ischemia as the underlying cause of cerebral palsy [MacLennan, 1999], a widely accepted, evidence-based standard for determining when cerebral palsy is due to hypoxia-ischemia is still lacking.
Some investigators prefer the term neonatal encephalopathy to describe the clinical syndrome of perinatal asphyxia because it is an all-embracing term that does not imply a single underlying etiologic disorder [Leviton and Nelson, 1992]. Signs and symptoms of brain dysfunction that constitute neonatal encephalopathy include low Apgar scores, failure to initiate and/or maintain respiration, depressed consciousness, abnormal tone (usually flaccidity), depression of developmental reflexes, and seizures during the first 48 hours of life. The clinical syndrome of neonatal encephalopathy may be preceded by a variety of prenatal and intrapartum risk factors [Badawi et al., 1998b], and usually is associated with acute brain injury occurring around the time of birth [Cowan et al., 2003].
Signs and symptoms of birth asphyxia or neonatal encephalopathy are strongly predictive of cerebral palsy in the child. For instance, term infants whose immediate postpartum course was comprised of a 5-minute Apgar score of 5 or less, continuing neurologic abnormalities, and seizures in the first days of life constitute a group at high risk for chronic motor disability (55 percent) and for death or disability combined (70 percent) [Ellenberg and Nelson, 1988]. The risk of cerebral palsy is increased 30-fold in infants with a 5-minute Apgar score of less than 7, and 80-fold in infants with a 5-minute Apgar score of 3 or less [Moster et al., 2001; Thorngren-Jerneck and Herbst, 2001; Wu et al., 2003].
Neonatal stroke, which encompasses ischemic perinatal infarction and sinovenous thrombosis occuring in the perinatal period (before the age of 7 days) or neonatal period (before 28 days of age), is a particularly important cause of cerebral palsy [Lynch and Nelson, 2001]. Ischemic perinatal stroke may be responsible for 28–50 percent of all cases of hemiplegic cerebral palsy in term infants [Humphreys et al., 2000; Uvebrant, 1988; Wu et al., 2003; Bax et al., 2006]. Among children with cerebral palsy who are referred for neuroimaging, 13–37 percent are diagnosed with a neonatal stroke [Ashwal et al., 2004]. Newborns with ischemic perinatal infarction either may present acutely during the neonatal period, with neurologic symptoms such as seizures, or may be clinically asymptomatic until several months of age, when pathologic handedness or seizures are first noted [Golomb et al., 2001]. The risk of cerebral palsy after ischemic perinatal infarction is especially high among infants with delayed presentation; this finding is not surprising, given the fact that hemiparesis is the most common presenting symptom in this group [Golomb et al., 2001; Wu et al., 2004b]. Involvement of the internal capsule also portends a poorer motor outcome after ischemic perinatal infarction [Mercuri et al., 1999; Wu et al., 2004b].
Neonatal sinovenous thrombosis also can lead to cerebral palsy. In a population-based study of pediatric stroke in Canada, 43 percent of all cases of sinovenous thrombosis occurred during the neonatal period [deVeber and Andrew, 2001]. Risk factors for neonatal sinovenous thrombosis include systemic illness, polycythemia, coagulation abnormalities, and extracorporeal membrane oxygen (ECMO) therapy, and motor sequelae such as cerebral palsy were more common if sinovenous thrombosis led to development of venous infarction [deVeber and Andrew, 2001; Wu et al., 2002]. Periventricular venous infarction [Takanashi et al., 2003], intraparenchymal hemorrhage, and birth trauma also may lead to cerebral palsy. (Please refer to Chapter 18 for further discussion of birth trauma.)
Brain Injury Related to Prematurity
Preterm infants constitute a disproportionate share of the cases of children who develop spastic diplegia but can manifest any cerebral palsy subtype. Both intraventricular hemorrhage and white matter necrosis seen in periventricular leukomalacia may occur before or after birth. The main pathogenetic mechanisms underlying periventricular leukomalacia are hypoxia-ischemia and inflammation [Volpe, 2009; Dammann and Leviton, 1997]. Periventricular leukomalacia and intraventricular hemorrhage are reviewed in Chapters 17–19.
Developmental Abnormalities
Brain malformations originating from intrauterine maldevelopment may underlie the neurologic impairment seen in children with cerebral palsy. As a group, children with cerebral palsy have a higher incidence of congenital malformations both within and outside of the brain [Rankin et al., 2009]. Population-based studies of cerebral palsy suggest that 9–14 percent of affected children have a brain malformation [Croen et al., 2001; Wu et al., 2003]. The most common malformations in term children are cortical dysplasia/polymicrogyria, schizencephaly, and pachygyria/lissencephaly. Complex brain malformations are the most frequent category among preterm infants with cerebral palsy [Ashwal et al., 2004].
A genetic or metabolic disorder may be associated with a specific brain malformation that causes cerebral palsy. For example, children with Zellweger’s syndrome typically have polymicrogyria and other cortical malformations. Miller–Dieker syndrome is a known cause of lissencephaly. Glutaric aciduria type I may masquerade as dyskinetic cerebral palsy [Hauser and Peters, 1998], and arginase deficiency mimics diplegic cerebral palsy [Prasad et al., 1997]. Children with cerebral palsy who demonstrate either progressive decline or atypical features such as dysmorphisms, macrocephaly, or a strong family history should be tested for an underlying genetic or metabolic disorder.
Prenatal Risk Factors
A number of maternal conditions have been associated with an increased risk of cerebral palsy in the offspring. Intrauterine inflammation, or chorioamnionitis, has received increasing attention as a potential risk factor [Grether and Nelson, 1997; Leviton et al., 1999; Wu and Colford, 2000]. Epidemiologic studies suggest that maternal intrapartum fever, a clinical or histologic diagnosis of chorioamnionitis, and serologic markers of inflammation in the fetus all confer an increased risk of cerebral palsy. In term infants, chorioamnionitis, often diagnosed by the presence of intrapartum maternal fever, is a particularly strong risk factor for spastic quadriplegia [Grether and Nelson, 1997; Wu et al., 2003]. Maternal fever during labor also increases the chance that the neonate will have low Apgar scores and characteristics of neonatal encephalopathy [Badawi et al., 1998a; Lieberman et al., 2000]. It is hypothesized that the fetal inflammatory response that occurs in the setting of an inflammatory intrauterine environment is responsible for brain injury leading to cerebral palsy [Dammann et al., 2002; Nelson et al., 1998]. Yet the mechanisms by which intrauterine inflammation might cause cerebral palsy remain controversial. It is postulated that inflammation may interact with a hypoxic-ischemic insult to injure the newborn brain in some children with cerebral palsy. A study has indicated that, in the presence of fetal acidemia, chorioamnionitis has poor predictive value and does not portend additional risk for development of neonatal encephalopathy [Shalak et al., 2005]. It has also been postulated that exposure to neurotropic viruses, such as herpes group B viruses, may also increase the risk of cerebral palsy [Gibson et al., 2006a; Djukic et al., 2009].
Intrauterine growth restriction is associated with an increased risk of cerebral palsy, especially in term infants [Jarvis et al., 2003; Topp et al., 1996]. Infants who are large for gestational age also have been reported to be at higher risk [Uvebrant and Hagberg, 1992]. Prothrombotic abnormalities, including factor V Leiden, methylenetetrahydrofolate reductase (MTHFR) C677T, and presence of anticardiolipin antibodies, have been found with increased frequency among infants with cerebral palsy and perinatal arterial infarction [Gunther et al., 2000; Hagstrom et al., 1998; Gibson et al., 2005], though not all studies have confirmed these associations [Miller et al., 2006]. A history of infertility also has been found to be associated with an increased risk of neonatal encephalopathy, developmental delay, and cerebral palsy [Badawi et al., 1998b; Ericson et al., 2002; Stromberg et al., 2002].
The pathogenesis of cerebral palsy is complex and multifactorial. For instance, among infants with perinatal ischemic infarction in the newborn period, multiple risk factors often are observed [Gunther et al., 2000; Wu et al., 2002]. When signs of hypoxia-ischemia and markers of infection both are present in a newborn, the risk for cerebral palsy is markedly heightened [Nelson and Grether, 1998]. Studies of placental abnormalities in cerebral palsy also suggest that a combination of thrombotic and inflammatory findings confers the highest risk of adverse neurologic outcome [Redline and O’Riordan, 2000].
A number of toxins have been described as causing cerebral palsy, such as benzyl alcohol preservative and in utero alcohol exposure [Benda et al., 1986; Burd et al., 2003]. Ingestion of methyl mercury by pregnant women in the Minamata Bay disaster resulted in the birth of children who were spastic, microcephalic, and cognitively impaired. Congenital infections, often referred to as TORCH infections (toxoplasmosis, rubella, cytomegalovirus, and herpes simplex infection), also can infect the fetus and produce serious encephalitides with motor sequelae [Sever, 1985].
Recent literature suggests that the risk of cerebral palsy may be modified by relatively common genetic polymorphisms. Inherited cytokine polymorphisms, including several found in the tumor necrosis factor-alpha and mannose-binding lectin genes, have been associated with increased risk of cerebral palsy in term infants [Gibson et al., 2006b, 2008]. Furthermore, a functional promotor region polymorphism in the interleukin (IL)-6 gene has been associated with cerebral palsy in term infants, as well as with the extent of brain injury and gray-matter volume in premature infants [Wu et al., 2009; Harding et al., 2004; Reiman et al., 2008; Djukic et al., 2009].
Postnatal Brain Injury
Bilirubin toxicity is a well-known cause of dyskinetic cerebral palsy, and continues to be a significant problem, in spite of recent progress in management of hyperbilirubinemic neonates [Shapiro, 2003]. Although bilirubin levels below 25 mg/dL rarely may be associated with kernicterus [Soorani-Lunsing et al., 2001], more commonly, bilirubin levels greater than 30 mg/dL are responsible for this problem. Symptoms of kernicterus in a jaundiced newborn usually are present by the second or third day of life. The child becomes listless and sucks poorly. Fever may be present, and crying becomes weak. The Moro and deep tendon reflexes become difficult to elicit, and general muscle tone becomes decreased. After several weeks, tone increases, and the infant manifests extension of the back with opisthotonos and marked extension of the extremities [Van Praagh, 1961].
The full clinical pattern of kernicterus infrequently is present in a single affected patient. Signs and symptoms consist of choreoathetosis, dystonia, tremors, and rigidity. Upward gaze and, on rare occasions, horizontal gaze may be impaired. Sensorineural hearing loss is common. Mental retardation, microcephaly, and spasticity may be present. A clinical score for assessing bilirubin-induced dysfunction in newborns has been recommended [Johnson et al., 1999]. Preterm infants with athetotic cerebral palsy have relatively uniform features, similar to term infants with kernicterus [Okumura et al., 2009]. Unconjugated bilirubin damages mitochondria and is toxic to neurons and to astrocytes [Ostrow et al., 2004]. Microscopic examination reveals yellow granules that are found not only in neurons, but also in the interstitial tissue. These changes occur in the first few days after injury. Neurons are grossly pyknotic, and in children who live for longer periods, large areas of cell loss, gliosis, and demyelination are evident.
The primary management problem for children affected with kernicterus is the lack of provision of the multiple services needed by affected infants and, more importantly, the failure to recognize early at-risk infants and manage severe hyperbilirubinemia in an efficient and timely fashion [Johnson et al., 2009; Maisels, 2009].
Clinical Features and Diagnosis
The clinical findings often change with maturation; both severity and distribution may be altered. A child with cerebral palsy who has been hypotonic may become hypertonic. A number of infants with mild abnormalities subsequently demonstrate a decrease or, in some instances, a disappearance of motor dysfunction [Nelson and Ellenberg, 1982]. The clinical picture should not include evidence of progressive disease or loss of skills previously acquired. Any suggestion of progression of disability should result in expansion of the differential diagnosis to explain the progression. The history should focus on an attempt to identify a specific cause and should especially investigate familial or metabolic disease that might have implications for treatment or family counseling [Barabas and Taft, 1986].
Further Diagnostic Evaluation
A useful algorithm for the evaluation of the child with cerebral palsy has been published (Figure 69-1) [Ashwal et al., 2004]. An abnormality is documented on cranial magnetic resonance imaging (MRI; in 89 percent of cases) or computed tomography (CT; in 77 percent) in a large percentage of patients; therefore, all children with cerebral palsy should undergo a neuroimaging study, preferably MRI [Accardo et al., 2004]. Because of the reported low incidence of metabolic and genetic disorders among children with cerebral palsy, testing for these disorders is indicated only in children in whom the history or clinical examination includes atypical features, or if a specific diagnosis is not established with neuroimaging. Children with a brain malformation also warrant consideration for further testing to determine if an underlying genetic or metabolic disorder is present. The presence of microcephaly at birth, dysmorphic features, and congenital anomalies outside the nervous system suggests early developmental defects. Microcephaly is one of the most common brain malformations and is caused by mutations of a number of different genes [Ashwal et al., 2009; Mochida, 2009]. The high incidence rates for mental retardation (52 percent), epilepsy (45 percent), ophthalmologic defects (28 percent), speech and language disorders (38 percent), and hearing impairment (12 percent) [Ashwal et al., 2004] make it imperative that all children with cerebral palsy be screened for mental retardation, ophthalmologic and hearing impairments, and speech and language disorders; nutrition, growth, and swallowing also should be closely monitored. Unfortunately, although neuronal plasticity likely aids improvement in brain dysfunction, significant injury persists [Johnston, 2009].
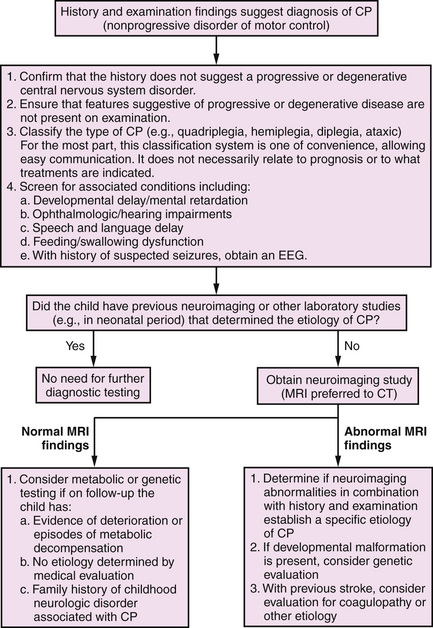
Fig. 69-1 Algorithm for the evaluation of the child with cerebral palsy (CP).
(From Ashwal S et al. Practice parameter: Diagnostic assessment of the child with cerebral palsy: Report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology 2004;62:851–863.)

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


