Figure 35-1 Brain magnetic resonance imaging scans in axial and coronal view, which demonstrate increased T2 signal in cerebral white matter, distributed diffusely and symmetrically throughout the centrum semiovale and sparing U-fibers and striatum. The changes are typical for a mitochondrial disorder.
At age 24, she was evaluated by a neurologist. She appeared very cachexic (height 5′7″, weight 90 pounds). Her mental status was normal. Cranial nerve examination revealed ptosis and restricted horizontal eye movements. Despite her lack of muscle bulk, her strength was normal. Sensory examination revealed decreased vibratory sensation in the fingertips and in a stocking distribution to the knees, but light touch and pinprick sensations were normal. Tendon reflexes were absent. The presence of Romberg’s sign, broad-based stance, and impaired tandem gait indicated sensory ataxia.
Electrophysiologic recordings (courtesy Dr. CF Bolton) from motor and sensory nerves of the upper and lower limbs indicated a severe demyelinating polyneuropathy: right median nerve motor conduction velocity (MCV) 24 m/s, distal latency (DL) 7.5 ms, compound motor action potential (CMAP) amplitude of the thenar muscle 9.4 mV, minimal F-wave latency 58.5 ms; right ulnar nerve MCV 24.8 m/s, DL 5.6 ms, CMAP of the hypothenar muscle 7.0 mV, minimal F-wave latency 65.3 ms; right peroneal nerve MCV (recording from tibialis anterior muscle) 13.6 m/s, DL 6.1 ms, CMAP 3.16 mV. Needle electromyography of the vastus medialis and tibialis anterior muscles revealed no spontaneous activity. Sensory nerve compound action potentials were not recorded with surface electrodes from the median, ulnar, and sural nerves. These findings corresponded with the signs of chronic demyelination and remyelination, and loss of axons observed in the prior sural nerve biopsy (Fig. 35-2). Routine laboratory studies revealed mild iron deficiency anemia and slightly elevated alanine transaminase (46 U/L; normal 4–26) and aspartate transaminase (99 U/L; normal 16–98). Cerebrospinal fluid (CSF) obtained by lumbar puncture revealed a normal cell count and elevated protein (124 mg/dL; normal <45).
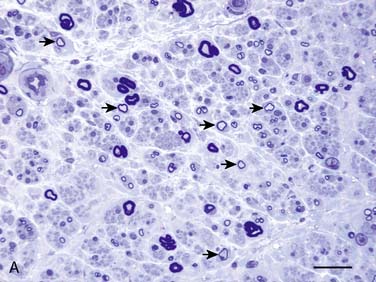
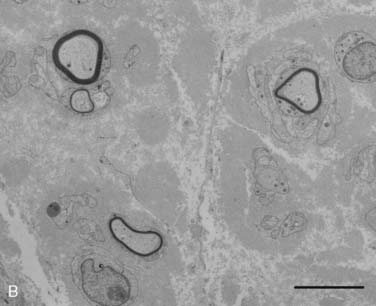
Figure 35-2 A, Transverse section of the sural nerve showing an overall reduction in myelinated fibers and many fibers with disproportionately thin myelin sheaths (arrows) indicating demyelination and remyelination; this occurs in the absence of inflammatory cells. Epon; Toluidine Blue; Bar 20 μm. B, Electron micrograph of sural nerve, which shows several thinly myelinated fibers surrounded by redundant Schwann cell cytoplasm, indicating demyelination and remyelination. Bar = 5 μm.
The patient’s demyelinating polyneuropathy and elevated CSF protein would be compatible with chronic inflammatory demyelinating polyradiculoneuropathy (CIDP); however, her other clinical manifestations, namely, prominent gastrointestinal dysmotility, cachexia, ptosis, ophthalmoparesis, and leukoencephalopathy led the neurologist to suspect MNGIE. A biopsy of the vastus lateralis muscle showed increased subsarcolemnal accumulation of mitochondria in several muscle fibers, with corresponding reduction of cytochrome c oxidase staining and increased staining with succinic dehydrogenase. Southern blot analysis and polymerase chain reaction amplification identified a predominant 7.5-kb mtDNA deletion as well as several smaller deletions in this muscle specimen.
Biochemical tests confirmed the diagnosis of MNGIE; thymidine phosphorylase activity in buffy coat was undetectable (normal mean ± standard deviation 634 ± 217 nmoL/h/mg-protein) and plasma thymidine was markedly elevated (6.4 μM, normal <0.05). DNA sequencing revealed compound heterozygous TYMP gene mutations, c.665A > G (p.222K>S) and c.1406insC (frameshift with premature stop codon).
The patient’s further disease course was marked by frequent hospital admissions due to episodes of vomiting, dehydration, and intractable abdominal pain caused by gastrointestinal pseudo-obstruction and associated bacterial overgrowth. Nutritional and vitamin requirements were maintained by regular TPN. The neuropathy continued to worsen to the point that patient became wheelchair-bound and had little use of her hands. She died at age 28 owing to a massive gastrointestinal hemorrhage.
Related posts:
 An Unusual Case of Pia-Arachnoiditis due to Metastatic Breast Carcinoma
An Unusual Case of Pia-Arachnoiditis due to Metastatic Breast Carcinoma
 Stepwise Facial and Upper Limb Weakness and Small Fiber Sensory Loss—Tangier Disease
Stepwise Facial and Upper Limb Weakness and Small Fiber Sensory Loss—Tangier Disease
 Late-Onset Transthyretin Val30Met Familial Amyloid Polyneuropathy Unrelated to Endemic Foci
Late-Onset Transthyretin Val30Met Familial Amyloid Polyneuropathy Unrelated to Endemic Foci
 Inflammatory Multifocal Encephalomyelopolyradiculoneuropathy Mimicking a Granulomatous or Lymphomatous Process
Inflammatory Multifocal Encephalomyelopolyradiculoneuropathy Mimicking a Granulomatous or Lymphomatous Process
 POEMS Syndrome
POEMS Syndrome
 Amyloidosis Typing Based on Laser Microdissection and Mass Spectrometry of Paraffin-Embedded Tissue Biopsies
Amyloidosis Typing Based on Laser Microdissection and Mass Spectrometry of Paraffin-Embedded Tissue Biopsies
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


