Fig. 8.1
A hairy patch found in the back of a child with congenital spinal deformity; it can be associated with occult spinal dysraphism. The presence of neurocutaneous stigmas is an unreliable indicator of intraspinal abnormality
Tethered spinal cord (Fig. 8.2a) is the most common MRI-identified intraspinal anomaly in congenital spinal deformity in many reports; syrinx (Fig. 8.2b) is the second; then thickened and fatty filum, low conus, diastematomyelia (Fig. 8.2c), intradural mass/lipoma, extradural mass, Chiari malformation arachnoid cyst, and Dandy–Walker malformation [18, 19, 23].
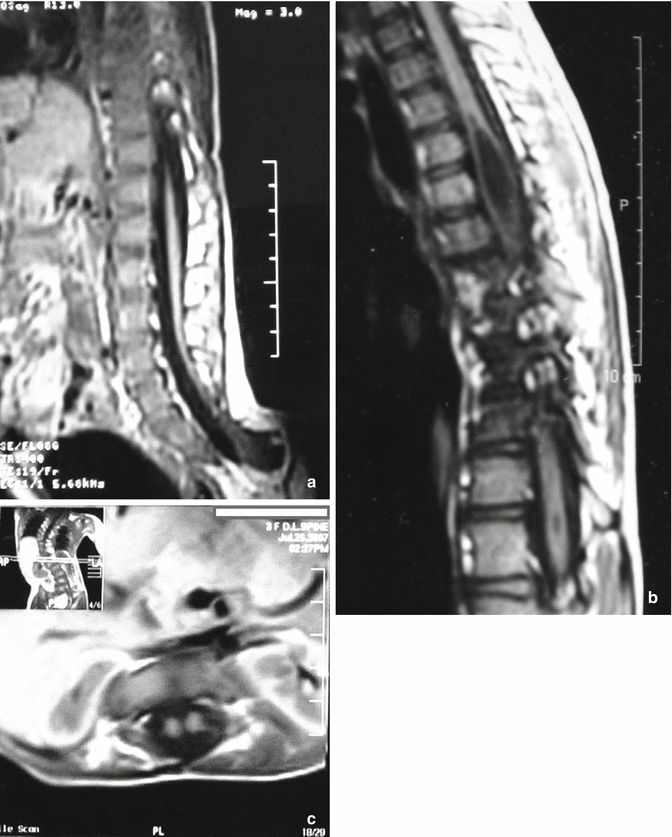
Fig. 8.2
Intraspinal anomalies associated with spinal deformities requiring neurosurgical consultation. (a) A sagittal T1-weighted MRI showing a tethered spinal cord, the most common MRI-identified intraspinal anomaly in congenital spinal deformity. Syrinx and tethered cord can also be found in case diagnosed as idiopathic scoliosis. (b) A sagittal T1-weighted MRI showing multiple variable sized dorsal syrinx associated with congenital scoliosis. (c) An axial T1-weighted magnetic resonance image (MRI) showing diastematomyelia with complete split of the cord at the dorsal region in a child with congenital scoliosis
Neurologic deficit caused by tethered spinal cord in congenital spinal deformity may not be manifested in very young children, and in older ages, there is lack of clear association between intraspinal anomalies and detectable clinical manifestations; therefore, MRI is generally recommended in the evaluation of patients with congenital spinal deformity even in the absence of clinical findings [23]. The performance of MRI in young children, especially 5 years of age or less, involves administration of sedation or general anesthesia, with the attendant risks of respiratory complications in these children who already may suffer from pulmonary compromise. As a result, a selective approach probably is wise. An MRI scan must be obtained in older age groups before surgical correction of the spinal deformity in cases with established or developing neurologic signs and probably also in cases with progressive deformity, in which surgery is to be considered sooner, but for younger children, MRI with the patient under general anesthesia is to be considered only if surgery is imminent or neurologic signs develop [18].
Marfan Syndrome
Dural ectasia is ballooning or widening of the dural sac, fibrillin deficiency resulting in connective tissue abnormality, and weakness in the dural sac has been suggested as the cause for duralectasia in Marfan syndrome. It usually occurs in the most caudal portion of the lumbosacral spinal column, at the point of greatest cerebrospinal fluid pressure in the upright patient. The neural symptoms are thought to be related to stretching and traction mechanisms, which may be clinically manifested with back pain and headaches. The consequences of duralectasia include bony erosion or anterior meningoceles. Widened interpediculate distance, increased vertebral scalloping, and increased sagittal diameter can detect duralectasia in patients with Marfan syndrome. Dural ectasia is a major diagnostic criterion used in Marfan syndrome, especially in patients who previously have not had sufficient major and minor diagnostic criteria [24, 25].
An incidence of duralectasia of 63 % was reported in Marfan syndrome [26]. This incidence was noted to be 76 % in patients with Marfan syndrome and back pain and 41 % in patients with Marfan syndrome without back pain [27]. Because the calculation of dural volume requires sophisticated software programs that are not widely available and because duralectasia is important as a major diagnostic criterion for Marfan syndrome, many guidelines were developed to detect the presence of duralectasia on computed tomography or MRI scan with trials to establish normative values for lumbosacral dural sac dimensions [28]. An abnormal dural sac ratio (the dural sac diameter corrected for vertebral size) has also been used to identify duralectasia in patients with Marfan syndrome. In symptomatic duralectasia, posterior laminectomy has been sometimes implemented as a means of relieving back pain.
Neurofibromatosis
Dystrophic neurofibromatosis scoliosis is characterized by early onset, rapidly progressive curves that are difficult to treat and has a tendency to progress to a severe deformity. Dystrophic curves may be associated with kyphosis and have a higher incidence of neurologic injury. Most of these patients present with skin lesions (Fig. 8.3) as well as associated neurofibromas that envelope the bone or come from the canal (dumbbell lesion) [29].
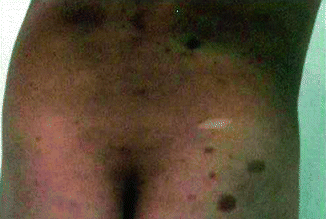
Fig. 8.3
Café au lait spots, cutaneous markers of systemic disorders observed in children with neurofibromatosis scoliosis
Enlargement of the spinal canal caused by intraspinal tumors or duralectasia is common. It erodes the bony and ligamentous structures causing vertebral scalloping and meningocele formation. Meningoceles, pseudomeningoceles, duralectasia, and dumbbell lesions are related to the presence of neurofibroma or abnormal pressure phenomena in and around the spinal canal neuraxis. Paraplegia is an uncommon finding in patients who have dystrophic curves; it is more prevalent in patients who have severe vertebral angulation (kyphosis), vertebral subluxation, and soft tissue tumors in the spinal canal [30]. Occasionally, these intraspinal elements may compromise the cord directly when instrumentation and stabilization are attempted, or they may cause erosive changes in the bone, preventing primary fusion. A rare, but important, cause of paraparesis in scoliotic patients is spinal cord compression due to rib penetration [23, 31]. A CT scan is the most sensitive tool to diagnose intraspinal rib dislocation. A resection of the rib will prevent or improve paraparesis in most patients who have dislocation.
It is the surgeon’s responsibility to correct and stabilize the spine with the most expedient, safe, and permanent method without causing neurologic injury. Therefore, it is imperative to evaluate such a condition in the preoperative period. MRI should be used in the investigation of all dystrophic curves before surgical treatment [32, 33].
Neuromuscular and Myelomeningocele Scoliosis
By definition, neuromuscular disorders are a group of diseases that affect any part of the nerve and muscle. These nerve tissue disorders include motor neuron diseases such as amyotrophic lateral sclerosis and spinal muscular atrophy, which may involve motor neurons in the brain, spinal cord, and periphery, and ultimately weaken the muscle. Many of these diseases can cause early onset scoliosis due to a primary affection in the neural axis.
Spinal deformity also may be caused by paralysis secondary to the spinal cord injury. Scoliosis is secondary to spinal cord affection in patients with very young age; traumatic paralysis of the spinal cord may also lead to syringomyelia or traumatic tethering in 20 % of patients and should be looked for when patients with spinal cord injury have worsening symptoms. Another type of scoliosis is defined by the presence of a clear anomaly in the spinal cord – “myelomeningocelescoliosis.” Both types of scoliosis, neuromuscular and myelomeningocele, have their own characteristics, complications, and way of management meriting a separate detailed discussion.
8.3.1.2 Cardiac
The relationship between cardiac abnormalities and scoliosis is a complex one. Both can originate from the same tissue defects in Marfan syndrome due to connective tissue disorder or in neuromuscular scoliosis due to different types of myopathy; scoliosis and congenital heart disease (CHD) can occur as a part of multiorgan congenital anomalies; in addition, there is an unexplained higher incidence of minor cardiac anomalies with idiopathic scoliosis. Conversely, scoliosis has a higher tendency to be present in children with congenital heart disease (CHD) with or without previous thoracotomy.
Idiopathic Scoliosis
Mitral valve prolapse (MVP) is known to be associated with thoracic skeletal anomalies, and MVP is four times more common in patients with severe idiopathic scoliosis than in the normal adolescent population. MVP and other valvular anomalies have been detected by echocardiogram and or ultrasound Doppler in 13.6–24.4 % of patients with idiopathic scoliosis as compared to 3.2 % in age- and weight-matched controls [14].
Patients with MVP are mostly asymptomatic, and only a systolic click or murmur can be detected on examination. Electrocardiogram (ECG) is abnormal in 21 % of patients with MVP as compared with only 1.6 % of patients with idiopathic scoliosis but no MVP. The persistent nature of MVP, even after corrective spinal surgery, may be related to factors other than geometric changes of the heart caused by abnormal thoracic curvature [12]. Looking at other comorbidities associated with idiopathic scoliosis, a significant relationship was found between valvular anomalies and other comorbidities. Valvular anomalies were detected in 17.2% with no comorbidity and in 50% with a comorbid condition; in this latter group of patients, routine echocardiography study seems advisable in the preoperative evaluation [34].
Congenital Scoliosis
CHD was found to be associated with congenital spinal deformity in 7–26 % of the patients. These include ventricular septal defects, atrial septal defects, patent ductus arteriosus, Fallot transposition of great arteries, pulmonary stenosis, sick sinus syndrome, and dextrocardia. Almost half of these children need medical therapy; some would require surgery for the cardiac condition in the future, and others will need to be kept under observation. This underscores the importance of a systematic clinical cardiac assessment and use of echocardiography for these patients. All patients in whom surgery is planned for correction of congenital spinal deformity should have echocardiography as part of preoperative workup. In addition, it has been suggested that patients with congenital scoliosis resulting from mixed bony defects and those with congenital kyphosis should have a routine echocardiogram because of the higher risk for CHD. These patients should be referred subsequently to a cardiologist for further management [18, 35].
Neuromuscular Scoliosis
Cardiac involvement may occur in most of the primary myopathies, including Duchenne muscular dystrophy (DMD), Becker muscular dystrophy (BMD), myotonic muscular dystrophy (MMD), and some cases of limb girdle muscular dystrophy. Dystrophin has been localized to the membrane surface of cardiac Purkinje fibers; this localization probably contributes to the cardiac conduction disturbances seen in DMD and BMD.
A high (60–80 %) occurrence of cardiac involvement is present in patients of all ages with DMD and BMD; this can be detected via ECG and echocardiogram. However, only about 30 % of patients with DMD have clinically significant cardiac complications. Pulmonary hypertension also has been implicated in the cardiorespiratory insufficiency associated with DMD, and some investigators blame congestive heart failure as the cause of death in as many as 40 % of patients with DMD. The cardiac compromise may be disproportionately severe relative to respiratory compromise in some patients with BMD. Thus, ECG and echocardiography screening are indicated at regular intervals for all patients with BMD because severe cardiac involvement in BMD may occasionally precede the clinical presentation of skeletal myopathy. Patients with myocardial involvement need close follow-up and treatment by a cardiologist with expertise in this area. Some patients with BMD may be suitable candidates for cardiac transplantation [36].
A high prevalence of abnormalities found via ECG exists in MMD. Studies have shown that about one-third of patients with MMD have first-degree atrioventricular block, while about one-fifth have left axis deviation. Only 5 % have left bundle branch block. Complete heart blockage, requiring pacemaker placement, is rare but can occur. Patients with MMD should receive routine cardiac evaluations [36, 37].
Marfan Syndrome
Marfan syndrome is characterized by connective tissue disorder with classic triad affection ocular, skeletal, and cardiac. Cardiovascular system anomalies account for a significant proportion of the shortened life span with Marfan syndrome.
The most prominent cardiovascular manifestations of Marfan syndrome are known to be caused by defects in fibrillin 1. MVP occurs in 35–100 %, aortic dilatation in 75 %, mitral regurge in 44–58 %, and aortic regurge in 15–44 %. Many patients present with silent MVP, diagnosed through echocardiography (78–100%), largely exceeding the auscultatory diagnosis (45–70 %). Therefore, it is recommended that all patients suspected for Marfan syndrome be evaluated echocardiographically [38], as progressive aortic root dilatation, aortic regurgitation, dissection, or rupture is the most common life-threatening feature. Aortic regurge is an indicator of high risk for subsequent complications such as dissection. In general, morbidity and mortality are associated with aortic abnormalities rather than with mitral valve dysfunction. Investigators reported that sporadic cases of Marfan syndrome have more severe cardiovascular involvement compared to familial cases [39].
Due to early diagnosis, the awareness for milder forms of the disease, advances in aortic surgery, and medical treatment, the life expectancy of Marfan patients has increased from 37 years in the seventies to more than 60 years in the nineties [38].
Congenital Heart Disease
It has been well established that the incidence of scoliosis is higher in patients with CHD than in normal subjects. The incidence of scoliosis in patients with CHD has been reported in the literature to be from 2 to 19 %. The relatively wide range in the incidence of scoliosis associated with CHD is thought to be due to differences in types of CHD, criteria of patient’s selection, effect of cardiac surgery, and the definition of scoliosis. In other words, the etiology of scoliosis associated with CHD is still unknown, and many factors such as CHD itself, cardiac surgery, thoracotomy, cyanosis, and other abnormalities may affect the onset of scoliosis. Some reports found strong correlation between thoracotomy done for cardiac surgery and the development of scoliosis in up to 22 % of their patients [39], whereas others found no correlation [40]. A number of theories have been proposed to explain the etiology of scoliosis associated with CHD, impaired oxygenation, and deficient or asymmetrical blood supply to the vertebral bodies or supporting tissues may be causative factors [41].
8.3.1.3 Urogenital
Congenital Scoliosis
The genitourinary and musculoskeletal systems are both of mesodermal origin and develop at the same time in the embryo. As a result, any genetic defect or other insult acting at a crucial stage of organogenesis, which results in a congenital vertebral abnormality, may also lead to a congenital genitourinary malformation. There is also the possibility that other developing organ systems will be affected. Thus, a cluster of disparate congenital abnormalities may occur. Renal anomalies are mostly nonhereditary, which supports the suggested etiology of an insult to the embryo between the fifth and seventh weeks. This period corresponds to the stage of organogenesis when the stem cell population is being established for the primordial organs. These interactions are sensitive to insult from genetic and environmental influences. In a 4-week-old embryo, the mesonephros is located from the sixth cervical vertebra to the lumbar spine. A stimulus in the lower cervical or upper thoracic area between the fourth and seventh weeks of gestation could simultaneously affect the developing mesodermal structures [42].
The frequent occurrence of congenital genitourinary abnormalities in patients with congenital scoliosis was reported in the 1980s [43]. The incidence of genitourinary abnormalities between 20 and 34 % has been found to occur in patients with congenital vertebral anomalies in different series using intravenous pyelography (IVP) and ultrasound. The most common urogenital anomalies associated with congenital spinal deformities are renal hypoplasia, horseshoe kidney, single kidney, congenital Megaureter, ectopic kidney (pelvic), hypospadius, pelviureteric junctional obstruction, posterior urethral valve, cloacal anomaly, epispadius, exstrophy of the bladder, hydronephrosis, and undescended testis [35]. It has been observed that the association of some malformation of the urinary system seems to be directly related to the occurrence of hemivertebrae; the location of the hemivertebra also seems important in relation to the side of agenesis of the kidney [43].
While these anomalies may remain asymptomatic, some can be associated with significant morbidity. Infection, obstruction, and the formation of calculus are the main reported problems. These patients are also at increased risk of proteinuria, hypertension, and renal insufficiency, and it is essential to have prolonged and careful follow-up. Some patients with urogenital anomalies (up to 25 % in some series) required surgery, others had abnormal renal function requiring medical therapy including dialysis, and the rest had abnormalities that do not affect renal function or do not require treatment [18].
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


