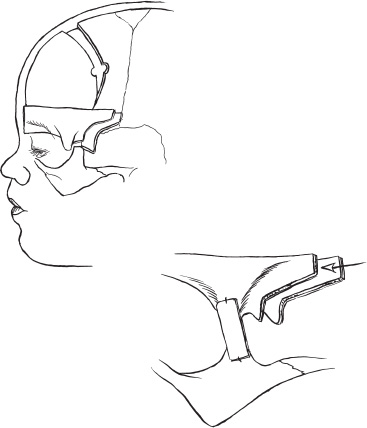
Surgical treatment of the various craniofacial syndromes has undergone a number of technical changes in the last several years. Various techniques, including strip craniectomies, lateral canthal advancement, facial augmentations, and partial to total calvarectomies, were developed to treat these congenital disorders. Over the last 15 years, our practice led us to use almost exclusively the bandeau/forehead reconstruction techniques developed by our French colleagues, Tessier, Marchac, and Renier. These techniques allow the surgical team to deal with the multiple anomalies that can occur in craniofacial syndromes, including restricted brain growth, asymmetric foreheads, and orbital dystopia. Craniofacial syndromes almost always involve a premature fusion/sclerosis of one or more cranial sutures. As a result, the patient can develop a number of craniofacial problems. If the coronal sutures are involved, typically a brachycephaly occurs, with fusion of both coronal sutures leading to a tall and flattened forehead. Retrusion of the orbital bandeau is not uncommon and usually causes a bandlike constriction over the orbits. If one side is more involved than the other, unequal growing planes result, leading the child to develop an orbital dystopia caused by an asymmetric orbital rims. In more severe syndromes, such as kleeblattschädel (cloverleaf skull), multiple sutures are affected and lead not only to facial and calvarial asymmetry but also to restriction in brain growth. To address the multiple problems seen in craniofacial syndromes requires correcting the facial asymmetry and orbital dystopia, allowing brain growth, and providing an aesthetic, symmetric alignment of the forehead and face. Typically, children with craniofacial syndromes are diagnosed at birth or at least by 3 months of age; the rare exception is oxycephaly, which is diagnosed at a later age (>3 years). The neonatalogist or pediatrician usually makes the first diagnosis. Once the craniofacial syndrome is recognized and diagnosed, a series of diagnostic studies can be done that are helpful in clarifying the operative approach to be taken. A routine skull series, in most cases, will identify the affected sutures and determine the presence of increased intracranial pressure (ICP), typically seen with the presence of thumbprinting or digital markings on the skull. Three-dimensional (3-D) reformatted computed tomography (CT) has proven extremely helpful in the preoperative planning as well as for documenting the extent of suture synostosis. Hydrocephalus is not uncommon in these disorders and also can be evaluated by CT scan. The presence or absence of a Chiari malformation also can be evaluated. The head circumference is measured from birth onward to assess head growth. It is not unusual to have restricted head growth in multiple suture closure. As part of the preoperative evaluation, the patient and family are seen by all members of the craniofacial team, which includes the neurosurgeon, plastic surgeon, pediatrician, pediatric neurologist, geneticist, social worker, and child-life specialist. As we have become more aware of the genetic influence in these syndromes, we involve the genetics team at the beginning to work up chromosome markers and to help with family counseling. After each team member has seen the patient and family, a conference is held, and the surgical recommendations to the family are planned. There has been much discussion over the years about the child who has both severe craniosynostosis and mid-face retrusion (e.g., in Crouzon’s syndrome) and the timing of the surgery to correct the calvarium and the midface. Ortiz-Monestario has been one of the strongest advocates for doing the monobloc advancement, in which the forehead and midface are both advanced early, that is, shortly after birth. Others have believed the risks of infection and blood loss and the severity of surgery to be too high and advocate doing the procedures in a two-stage fashion. The forehead and orbital regions are corrected shortly after birth, with the midface done later, at about age 8 to 9 years, depending on the facial maturity and presence of permanent dentition. The arguments for both sides are extensive and beyond the scope of this chapter. In craniofacial surgery, timing is extremely important. Several factors should be evaluated in making a decision about when to operate. Initially, we believed these children should be operated on as soon as possible, even as early as 2 to 3 weeks of age; however, we now prefer to wait until the child is 4 to 6 months of age to allow a more mature hematologic system and a larger blood volume for the anesthesiologist. In addition, the calvarial bone is firmer and has a more mature matrix, allowing better contouring and placement. The 6- to 12-month age period is critical in the developing child. This period is one of rapid head growth, and this growth assists in the final remodeling of the child’s head and face. Therefore, it is critical to use this period of rapid growth to assist in the reconstruction. In certain situations in craniofacial syndromes, however, premature suture fusion and restricted head growth have made it necessary to perform surgery earlier to allow adequate brain growth and to preserve vital brain function. In particularly severe cases [e.g., kleeblattschädel (cloverleaf skull)], we have done calvarial expansions as early as 2 weeks of age. This group of children not uncommonly (>80%) will need later surgery for aesthetic corrections; in addition, they have the propensity to restenose, again requiring a calvarial expansion. In children who present with severe cloverleaf deformities at birth, almost all (and in our experience all) children develop hydrocephalus early. CT is obtained for these children shortly after birth, usually within the first 48 hours. The presence of enlarged ventricles merits a rapid, early placement of ventriculo-peritoneal shunt. We elect to place the shunt through an occipital burr hole so that the anterior and middle portions of the skull are easily accessible for the later calvarial reconstruction. An additional helpful step is to elevate the pericranium as a second layer and place the shunt under this layer. If the pericranium is closed over the shunt, this will provide an additional layer of tissue to protect the shunt apparatus during later surgical reconstruction. Children with craniofacial syndromes often have other systemic problems that need to be evaluated. Routine ultrasounds are done of the organ systems to rule out any abnormality of the heart and kidneys. In children with multiple suture fusions, hypothalamic abnormalities also may be present; for this reason, routine endocrine examinations are done prior to surgery. We have the pediatrician do a well-baby checkup the day before the child is to be admitted to the hospital. If the child comes in with a fever and a workup reveals no active bacterial infection (normal white blood cell count with no shift), we proceed with the surgery. If the child has a upper respiratory infection but no signs of congestion, we also will proceed. If the child appears septic or an active bacterial source is identified, surgery is cancelled and rescheduled. Because these children often require blood transfusions, we offer the family the opportunity to provide donor-directed blood. Our experience is that more than 80% will take advantage of this option. In the child who presents with multisutural synostosis and marginal signs of increased ICP, a helpful adjunct test is ICP monitoring. The work by Marchac and Renier has been extremely useful in determining the ICP levels that are clinically significant. Their work has shown that children with ICP readings greater than 15 mm Hg merit serious consideration for a calvarial expansion. In our center, we admit the child for a 2-day inpatient stay in the intensive care unit (ICU). A lumbar drain is placed at the L4–L5 level, left in situ, and connected to a transducer. A graph is kept at the bedside with readings taken and recorded every half hour. Notes are kept of when the child is awake or asleep and during rapid eye movements (REM) sleep. Acceptable pressure readings are up to 10 mm Hg, ranges of 10 to 15 mm Hg are suspect, and persistent readings of greater than 15 mm Hg are consistent with abnormal ICP. An alternative technique for measuring ICP is using an epidural transducer placed through a small trepanation between the dura and skull. The same criteria for length and timing apply here as for measurements. General anesthesia with paralytic agents is used in all cases. Inhalation agents that increase ICP are avoided. Because of potential movement of the head, the airway must be secure, and because of potential blood loss, all patients require at least two large-bore intravenous 20-gauge lines or larger. Arterial lines are placed for monitoring blood gases, hematocrit, and electrolytes during the procedure. A Foley catheter is placed to monitor urine output. We do not routinely use steroids or anticonvulsants. Antibiotics are used (oxacillin 50 mg/kg) beginning with a preoperative dose and carried out for 24 hours. Our anesthesiologists will volume-load the child with crystalloid solutions at the beginning of the case. This hemodilutes the blood, thus helping to reduce blood loss. “Tanking up” the patient also reduces the risk of air embolism. In children with severe cloverleaf anomalies, in particular those with Pfeiffer’s syndrome, the cranial-base hypoplasia and midface anomalities cause significant airway obstruction. These children are typically obligate oral-airway breathers. For children with midface retrusion, abnormal palates, and large tongues, a tracheostomy is sometimes placed, usually within the first week of life, to prevent airway obstruction. Children with severe cloverleaf deformities typically have shallow orbits and, as a result, present with severe orbital proptosis. An early oculoplastic consultation is extremely important for discussion about how to protect the globes and prevent keratopathies. Tarsorrhaphy, conjunctival flaps, and other measures are often necessary to protect the globes until the forehead and midface advancements can be accomplished. The patient is placed in a supine position with the head resting in a horseshoe headrest. In cases where a total calvarial removal is planned, the head is flexed more forward, and the U-shaped headrest is used in place of the horseshoe. Some surgeons prefer the sphinx position, in which the child is placed prone and the head is hyperextended, with the child resting on a bolster or beanbag. If this position is to be used, preoperative radiographs of the cervical spine need to be done to rule out any congenital abnormalities. This position is contraindicated in the child with a Chiari malformation. The draping is done so that the head is fully exposed from the nasal tip to vertex to inion. A 180-degree access to the head and facial region is required so that no stands are placed to either side of the patient’s head. The anesthesia team is placed parallel to the patient’s side at foot level. The nursing team is placed on the opposite side at the foot of the patient. A small Mayo stand is placed over the patient’s abdomen and a second mobile stand is placed to the side for surgical trays. Because multiple teams are involved in a staged fashion, several surgical trays are needed. Our nursing team has found it beneficial to keep a large table in the background to hold the various tray setups. As each surgical team comes into the field, their instruments are placed on the mobile table, and the required working instruments are placed on the Mayo stand. A bicoronal incision is performed and carried from ear to ear behind the hair line. Over the last several years, we switched to the stealth, or zigzag incision. Making several curves in the incision reduces the hair parting over the incision when the hair is wet and also seems to reduce the hypertrophic scarring that occurs over the temporalis muscle. In most children, it is not necessary to shave the hair; instead, the incision can be carried through a parted hairline. In some cases, hair is shaved at a width of about 1 cm to allow placement of the incision and convenience in the later closure. The skin flap is elevated separate from the pericranium and carried down to the orbital rims bilaterally. Both frontozygomatic sutures must be exposed. In cases where a total calvariectomy is to be performed, the posterior flap is carried down to the inion. The pericranium then is elevated as a second layer and also carried down to the orbital rims. The neurovascular bundle is identified at the supraorbital notch and opened. There is often a thin rim of bone over the notch that can be easily opened with an osteotome. The dissection then is carried further around and under the orbital rims. At completion of the skin incision and forehead flap, both orbital rims and frontozygomatic sutures are exposed. Posteriorly, the inion and lambdoid sutures are exposed. The temporalis muscles are elevated from their insertion point at the temporal line down to the level of the zygoma. The belly of the muscle is not incised because to do so would cause atrophy and later a cosmetic deformity (“dimpling” in the pterion region). The temporalis muscle is elevated using a monopolar needle tip cattier, winged out on its base in what I call a “fish-belly” fashion. The orbital rims are dissected further until the nasion suture is exposed. Laterally, the orbital rims are dissected to the attachment of the lateral canthal ligament. We rarely detach this ligament except in severe cases of orbital dystopia. If the ligament is to be detached, an identification suture is placed through the ligament and then cut on the side closest to the orbital wall. This suture is helpful for locating and reattaching the ligament at the end of the procedure. We use two types of reconstruction in surgically correcting craniofacial anomalies. In children with only brachycephaly, we elevate the original orbital bandeau and then reshape it prior to replacement. The bandeau should be over advanced at the initial placement to allow for adequate brain growth. If placed in its normal position, the child will rapidly develop a “waistbanding” or constriction just over the orbits. In cases where the bandeau is too deformed, a new bandeau is harvested from over the calvarial vertex. In a brachycephalic child or one with a cloverleaf deformity, the forehead unit is almost always deformed. To correct for this, a new forehead is marked out with a Marchac template. Figure 6–1 shows the osteotomy cuts; intraoperatively, these will be marked out with methylene blue. The team first makes the decision as to which type of craniotomy (partial or total calvariectomy) is to be performed, and then the appropriate marks are mapped.
SURGICAL INDICATIONS AND PREOPERATIVE EVALUATION
Timing of Surgery
Hydrocephalus
PREOPERATIVE MANAGEMENT
Intracranial Pressure Monitoring
INTRAOPERATIVE TECHNIQUES
Anesthetic Techniques
Airway Management
Ophthalmological Considerations
Operative Position
Initial Exposure: Skin Incision and Flap Elevation
Craniotomy and Craniofacial Reconstruction
![]()
Stay updated, free articles. Join our Telegram channel


Full access? Get Clinical Tree


