Chapter 91 Diseases of the Neuromuscular Junction
Both acquired and inherited disorders of the neuromuscular junction (NMJ) are seen in childhood. As with adults, the most common NMJ disorders are autoimmune and respond to immunosuppressive therapy. These include myasthenia gravis (MG) and, rarely, the Lambert–Eaton myasthenic syndrome (LEMS). A number of genetically determined disorders of neuromuscular transmission, the congenital myasthenic syndromes, are unique to childhood and are discussed later in this chapter. Botulism, a toxin-mediated disorder of the NMJ, most commonly occurs in infancy. All NMJ disorders have the ability to produce generalized weakness and fatigability with a propensity for oculobulbar involvement. Electrophysiological studies will detect an impairment of neuromuscular transmission in most of these disorders [Harper, 2004]. Fortunately, most of these disorders are treatable [Barohn, 2008]. Before descriptions of the various NMJ disorders are given, normal relevant physiology is reviewed.
The Neuromuscular Junction
Familiarity with the pathophysiology, diagnosis, and treatment of NMJ disorders requires a fundamental understanding of normal neuromuscular transmission (Figure 91-1). Acetylcholine is the natural neurotransmitter of the NMJ. It is synthesized and stored in vesicles in the motor nerve terminal [Drachman, 1978]. Each vesicle contains a quantum (about 10,000 molecules) of neurotransmitter. At rest, individual vesicles spontaneously release their quantum of acetylcholine at special release sites on the presynaptic membrane. The released neurotransmitter migrates and binds to acetylcholine receptors (AChR) located on the postsynaptic membrane, producing a transient increase in the permeability of the membrane to sodium and potassium ions. The local end-plate depolarization that results is known as a miniature end-plate potential (MEPP).
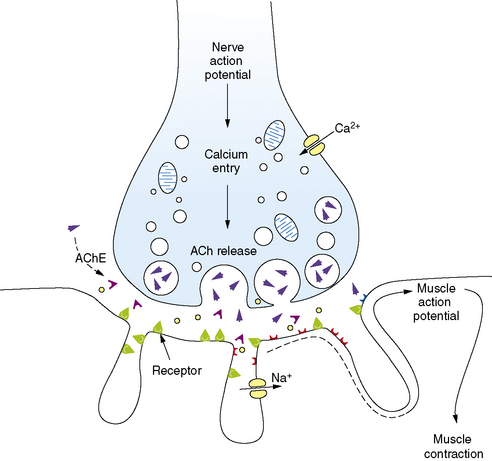
The amplitude of the EPP is directly related to the number of acetylcholine molecules that bind to their receptors. Normally, as a result of what has been termed the “safety margin” of NMJ physiology, the released amount of neurotransmitter is more than sufficient to produce a muscle action potential [Hughes et al., 2004]. As discussed later, the immunologic defect in the NMJ disorder, myasthenia gravis, directly reduces this safety margin, and muscle weakness ensues. A change in safety margin is also seen in other NMJ disorders.
Neuromuscular transmission is rapid, taking only milliseconds to complete the entire sequence. The process is terminated by diffusion of acetylcholine from the synapse and its rapid hydrolysis by acetylcholinesterase (AChE) [Lui and Erickson, 1994].
By 20 weeks’ gestation, the structure of the NMJ is well established, with further refinement of the postsynaptic membrane continuing until term [Sanes and Jessell, 2000]. Clustering of AChR in utero is dependent on muscle-specific receptor tyrosine kinase (MuSK) and rapsyn. Both the AChR and MuSK have been implicated as postsynaptic targets of the immune system in MG [Hoch et al., 2001). Other NMJ antigens targeted by plasma factors are likely to be discovered in the future [Plested et al., 2002].
Autoimmune Myasthenia Gravis
MG is the best-understood autoimmune disease of the nervous system [Vincent et al., 2001]. The immune-mediated nature of MG was suspected as early as 1960, when Simpson speculated that it was an autoimmune disease with antibodies directed against skeletal muscle AChR [Simpson, 1960]. A series of breakthroughs in the 1970s confirmed Simpson’s hypothesis. Lindstrom and colleagues developed the animal model experimental autoimmune MG by immunizing rabbits and rats with highly purified AChR from the electric organ of the eel [Patrick and Lindstrom, 1976a; Lindstrom et al., 1976b]. Not only did the animals develop weakness and respiratory insufficiency in about 3 weeks, but they responded to anticholinesterase medications and showed typical decrementing responses on repetitive nerve stimulation [Seybold et al., 1976]. These animals also had high AChR antibody titers in the serum. Subsequently, these titers were found in the serum of MG patients [Lindstrom et al., 1976]. Engel and co-workers localized both the IgG antibody and complement to the myasthenia motor end plate [Engel et al., 1977; Engel and Arahata, 1987]. This implied that circulating immunoglobulin (Ig) G antibody directed against the AChR bound to the postsynaptic membrane and activated the terminal complement sequence (C5b-9), or membrane attack complex, resulting in lysis of the AChR with subsequent degeneration.
Elevated membrane attack complex levels have been demonstrated in the plasma of MG patients [Barohn and Brey, 1993]. Anti-AChR antibody has also been shown to block neuromuscular transmission and accelerate turnover of AChR cross-linked by IgG [Drachman, 1978]. As a result of this process, the postsynaptic membrane becomes simplified with decreased junctional folds [Engel et al., 1976]. In addition, the neuromuscular blockade is passively transferred by injecting animals with IgG from MG patients [Toyka et al., 1977]. The same phenomenon occurs when an infant born to a mother with MG exhibits symptoms at birth, so-called neonatal MG [Papazian, 1992˜].
The AChR is a large protein consisting of five subunits, and the antibody response in MG and experimental autoimmune MG is polyclonal. The portion of the protein primarily responsible for inducing antibodies that produce the disease is debated. Although the existence of a main immunogenic region in the alpha subunit has been promoted [Tzartos and Lindstrom, 1980], other investigators have challenged this evidence [Lennon and Griesmann, 1989]. It is possible that, if the most pathogenic determinants of the AChR can be identified, a more rational and specific immune therapy can be designed. Multiple mechanisms are known to cause loss of functional AChRs in MG; these include complement-mediated lysis, accelerated internalization and degradation of AChRs, and direct blockade of AChRs by antibodies [Drachman et al., 1980; Meriggioli and Sanders, 2009]. Among these, complement-mediated lysis is thought to be the most important mode of loss of AChR.
The process that initiates the immune-mediated NMJ dysfunction is still unknown. The thymus gland may play a role, as 75 percent of MG patients who undergo thymectomy have thymic pathologic findings, with 15 percent being tumors of the thymus and the remainder consisting of lymphoid hyperplasia [Castleman, 1966]. Lymphocytes in the thymus and peripheral blood appear to be sensitized to muscle in MG patients [Sommer et al., 1990, 1991]. Muscle-like myoid cells are found in the thymus gland, and thymus tissue from MG patients with and without thymoma is enriched in AChR-reactive T cells [Wekerle et al., 1975; Kao and Drachman, 1977]. The close association of lymphocytes and myoid cells in the thymus, along with some stimulus causing the disruption of immune tolerance, may lead to the autoimmune response. There may be a hereditary predisposition to develop MG because there is an increased incidence of certain human leukocyte antigens (HLA) in various MG populations [Meriggioli and Sanders, 2009].
Epidemiology
MG has a prevalence of approximately 125 cases per million population [Drachman, 1994]. Approximately 11–24 percent of all MG patients have disease onset in childhood or adolescence [Simpson, 1958; Millichap and Dodge, 1960]. There is a slight female predominance of 3:2, although males predominate in older age groups. The disease can arise at any age, but peaks are observed in the third and sixth decades.
Clinical Features
The best data on the natural history of MG are from Grob and co-workers, who carefully studied 1976 patients between 1940 and 2000 [Grob et al., 2008]. Some of the key points from their work follow:
Clinical Classification
Osserman classified MG patients according to disease severity [Osserman, 1958]. The most commonly used modification of the original classification is as follows:





The Osserman classification has several shortcomings, including the vague descriptive terminology and lack of distinctions between some groups. For instance, differentiating between “mild” and “moderate” generalized disease may be difficult. The scheme also fails to include a category for patients in remission. A task force of the Myasthenia Gravis Foundation of America (MGFA) developed a new classification system (Box 91-1) that is more descriptive and provides better distinction between classes [Jaretzki et al., 2000; Barohn, 2003].
Box 91-1 Myasthenia Gravis Foundation of America Classification System

Class II



Class III



Class IV




Categories of Myasthenia Gravis in Childhood
Autoimmune MG in children is most commonly divided into neonatal transient and juvenile types.
Neonatal transient MG occurs in infants of myasthenic mothers. Placental transfer of anti-AChR antibody or immunocytes results in transient impairment of neuromuscular transmission in the neonate [Barlow, 1981; Donaldson et al., 1981]. Neonatal MG has also been identified in mothers who have MuSK antibodies [O’Carroll et al., 2009]. Findings such as a weak suck or cry, ptosis, dysphagia, generalized weakness, decreased spontaneous movement, or respiratory distress are usually present in the first few hours of life but may not be evident until the third day [Millichap and Dodge, 1960]. The majority of patients have hypotonia or transient weakness and a very small proportion present with arthrogryposis multiplex congenita [Jeannet et al., 2008]. The hypotonia and transient weakness usually resolve in the first 4 weeks but may persist for months [Desmedt and Borenstein, 1977; Branch et al., 1978], and sometimes can lead to persistent facial and bulbar manifestations [Jeannet et al., 2008]. The severity of the disorder in the infant does not correlate with the degree of maternal involvement, but there is evidence that a higher maternal antibody titer may predict severity and onset of neonatal myasthenia [Eymard et al., 1989]. A prior history of neonatal MG in a sibling is the only predictive factor. Fortunately, only 10–15 percent of infants born to myasthenic mothers develop the disorder [Fraser and Turner, 1953; Namba et al., 1970; Ahlsten et al., 1992]. Prior thymectomy or remission of disease in the mother does not prevent development of neonatal transient MG [Geddes and Kidd, 1951; Elias and Appel, 1979], but has decreased the likelihood of neonatal myasthenia [Djelmis et al., 2002]. Careful monitoring of pregnant women with MG is critical because there is a 40 percent chance of disease exacerbation during pregnancy and a 30 percent risk in the puerperium. Perinatal mortality is approximately 68 per 1000 births, five times the risk in uncomplicated pregnancies [Plauche, 1991].
Juvenile MG represents the childhood onset of autoimmune MG seen in adults, but there are differences in the presentation [Chiang et al., 2009]. Onset is usually after 10 years of age, and disease manifestations appear before puberty in half the cases. Onset before 1 year of age is exceptional [Fenichel, 1978; Andrews et al., 1993; Geh and Bradbury, 1998]. Pubertal status might affect the clinical presentation, with higher incidence of ocular MG in prepubertal patients and generalized MG in postpubertal patients [Batocchi et al., 1990; Evoli et al., 1998]. Asian children have a higher incidence of ocular presentation [Chiang et al., 2009]. Female predominance was observed only after the age of 10 years [Haliloglu et al., 2002].
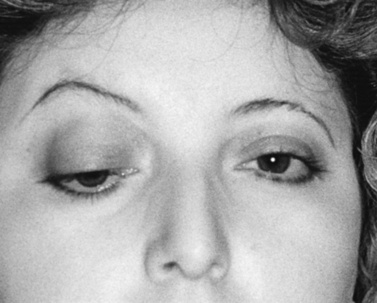
As with adults, ptosis is the most common clinical finding, frequently accompanied by ophthalmoparesis (Figure 91-2). Ptosis was unilateral at onset in one-third of juvenile MG patients, but subsequently spread to the other eye in nearly 90 percent of cases [Afifi and Bell, 1993]. Facial and oropharyngeal weakness is another common finding, producing dysarthria, dysphagia, and difficulty chewing. Facial weakness without ocular involvement is an unusual but recognized presentation of juvenile MG [Kini, 1995]. Extremity weakness can occur and is usually most prominent proximally. Bulbar weakness, characterized by slow chewing, dysphagia, nasal dysarthria, and weak cough, develops in up to 75 percent of patients [Rodriguez et al., 1983]. Respiratory failure from either diaphragmatic or intercostal muscle weakness or airway compromise related to bulbar dysfunction produces myasthenic crisis, an exacerbation severe enough to endanger life. As in adults, the disease may be generalized at onset, but isolated ocular involvement is a more common presentation, followed by generalization at a later time. However, children with ocular MG appear more likely than adults to remain with purely ocular disease. As many as 85 percent of adults with ocular MG later go on to develop generalized disease [Weinberg et al., 1994; Evoli et al., 1998]. In children, this percentage is closer to 50–75 percent [Afifi and Bell, 1993; Andrews, 2004]. Different HLA antigens are linked to various ethnic groups. Asian children tend to have HLA DRw9 [Wong et al., 1992], Caucasians are DQ8- or DR3-positive, and patients of African descent tend to have DR5 [Christiansen et al., 1984].
MG is frequently associated with other diseases, specifically those with an immune etiology. The most common are rheumatoid arthritis, thyroid disease, systemic lupus erythematosus, and diabetes mellitus [Millichap and Dodge, 1960; Rodriguez et al., 1983; Afifi and Bell, 1993]. Nonimmune disorders associated with juvenile MG include epilepsy in 3–13 percent [Snead et al., 1980; Rodriguez et al., 1983], various forms of neoplasia, particularly thymoma, and, later in life, breast carcinoma. Thymoma in juvenile MG, present in less than 5 percent of children Rodriguez et al., 1983], is relatively rare compared to adults and is mainly found in children with teenage onset [Andrews, 2004].
Neurologic Examination
Testing for weakness in the orbicularis oculi muscle is critical but often overlooked. Many symptomatic MG patients have bilateral weakness of this muscle group. Strength should also be tested in the lower facial muscles (blowing out cheeks against resistance) and in the tongue. Attention to speech patterns may disclose a nasal dysarthria. It is important to check for neck flexion and extension weakness because these muscle groups are frequently involved. Testing of extremity strength should include proximal and distal muscle groups in the arms and legs. Proximal limb muscles tend to be more affected than distal muscles. Rarely, MG patients may demonstrate a predilection for weakness in distal muscle groups, especially finger extensors [Nations et al., 1999].
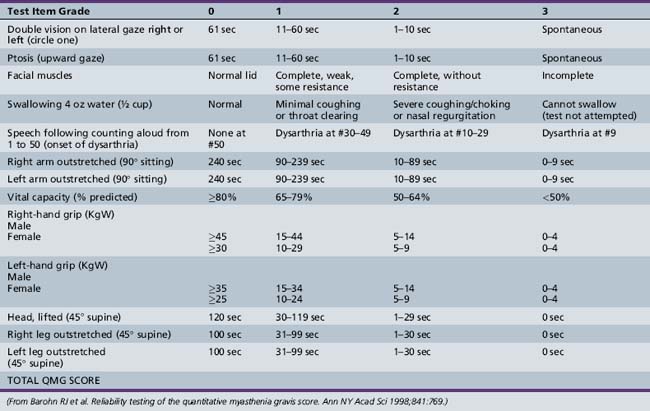
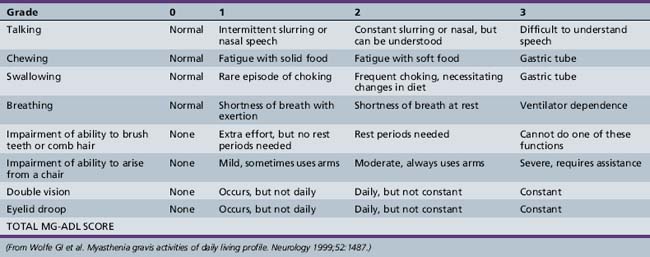
Symptoms and signs can be quantified by using a validated MG scoring system and activities of daily living scale (Tables 91-1 and 91-2). The activities of daily living scale correlates with the objective quantitative MG score [Wolfe et al., 1999]. An MGFA task force has recommended that the quantitative MG score be used as an outcome measure for therapy trials in MG [Barohn, 2003]. The scales are readily administered to adolescents as young as 15 years of age [Barohn et al., 1998]. A newer composite MG score with 10 items has recently been validated in adults [Burns et al., 2008] and it should be equally easy to administer in children as it was in adults.
Clinical and Laboratory Tests
Edrophonium (Tensilon) Test
The intravenous administration of up to 10 mg of edrophonium is often the first diagnostic test performed in the evaluation of a potential MG patient. However, the edrophonium test has a number of pitfalls. The most common mistake is that the physician performing the test does not have an objective parameter to measure before and after edrophonium administration. The most useful parameter is the degree of ptosis in each eye. The best indication of a positive test is a significant increase in the palpebral fissure aperture or the opening of a completely ptotic eye. If no ptosis is present, the edrophonium test may be difficult to interpret even in clear-cut cases of MG. If the patient has a severe restriction of extraocular movement and edrophonium dramatically improves the motility, the test is considered positive. However, subjective diplopia may not resolve unless edrophonium produces orthophoria in the eyes, which is rare. Significant improvement in dysarthria or in swallowing is another indication of a positive edrophonium test. A mild improvement in limb strength or subjective well-being is not sufficient to claim a positive test. In addition, a positive edrophonium test is not specific because transient subjective improvement is reported in other neurologic disorders, such as motor neuron disease and peripheral neuropathy [Oh and Cho, 1990].
The edrophonium test is performed in a straightforward manner. First, 1 mL (10 mg) of edrophonium is drawn up into a 1-mL tuberculin syringe. The edrophonium is often injected directly into a vein (usually antecubital), but in smaller children a “butterfly” intravenous line can be started, connecting the tuberculin syringe directly to the intravenous line. In children weighing less than 30 kg, the total delivered dose should not exceed 0.1 mg/kg. For children weighing more than 30 kg, a total dose up to 0.2 mg/kg may be given. Initially, a test dose of 0.01 mg/kg is injected. If, after 30–60 seconds, the patient experiences no side effects from the drug (fasciculations, sweating, nausea), further aliquots of 0.01–0.02 mg/kg are injected, not exceeding the total dose suggested. When injecting into an intravenous line, line flushes with saline are needed with each aliquot. Compared with adults, children are more likely to experience nausea, so an emesis basin should be available. More serious side effects, such as bronchospasm or lightheadedness caused by bradycardia, are quite uncommon, but atropine should be readily available. When either side effects or a positive response are obtained, no further edrophonium need be given. The mean dose of edrophonium needed to produce a positive response was 3.3 ± 1.6 mg for ptosis and 2.6 ± 1.1 mg for oculomotor dysfunction in a survey of 83 adult patients [Kupersmith et al., 2003].
In infants and younger children who are uncooperative and difficult to monitor over brief time periods, longer-acting neostigmine may be preferred. The intramuscular dose is 0.15 mg/kg and the intravenous dose is 0.05 mg/kg [Andrews, 2004]. Intravenous use can be hazardous due to severe muscarinic side effects [Wolfe et al., 1997]. A positive response is generally evident by 15 minutes and is most obvious after 30 minutes. Positive results on edrophonium or neostigmine testing are seen in up to 90 percent of juvenile MG cases [Afifi and Bell, 1993]. It is a good idea to have injectable atropine available in the case of severe side effects whenever using neostigmine.
Electrophysiologic Testing
Repetitive stimulation
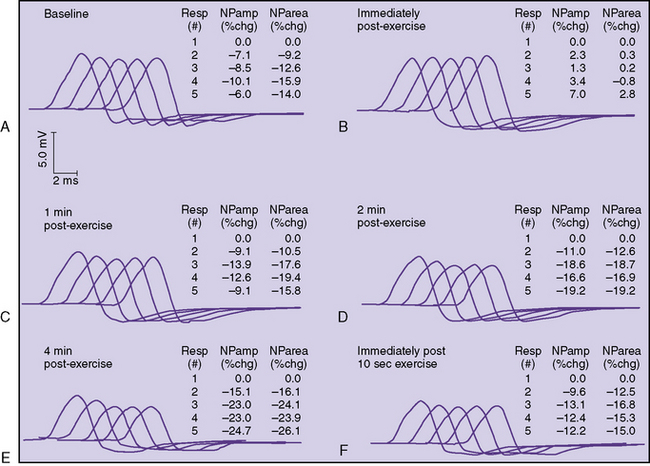
The classic electrophysiologic demonstration of an NMJ transmission defect is the documentation of a decremental response of the compound muscle action potential (CMAP) to repetitive stimulation (RS) of a motor nerve [Oh, 1988]. The decrement is due to failure of some muscle fibers to reach threshold and contract when successive volleys of ACh vesicles are released at the NMJ. Failure to reach the threshold EPP to achieve muscle contraction is called blocking. The percentage decrease in amplitude and area is calculated between the first CMAP produced by a train of stimuli and each successive one. In most laboratories, five or six responses are obtained at 2 or 3 Hz, and the maximal percentage decrement can be measured at the fourth or fifth response. A decrement of greater than 10 percent is considered a positive RS study (Figure 91-3).
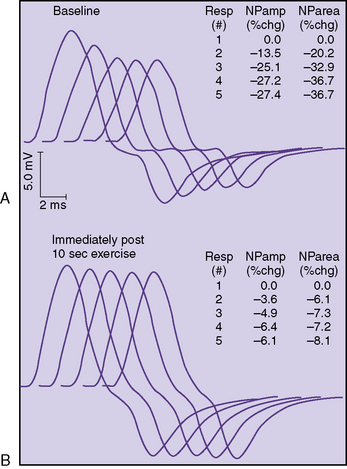
In some patients, a decremental response can be demonstrated at baseline. However, often a brief period of exercise (usually 1 minute) is required to fatigue the NMJ so that the decrement can be observed. This phenomenon of postexercise exhaustion (PEE) usually occurs at 2–4 minutes after exercise (Figure 91-4). In addition, repair or an improvement in the decrement can sometimes be observed immediately (within seconds) after brief exercise (see Figure 91-3).
A protocol for RS of the ulnar nerve recording over the adductor digiti minimi follows:







A decremental response is more likely present in a proximal muscle than in a distal muscle. In the series by Stalberg and Sanders, a decrement in a distal muscle was reported in 38 percent of patients, whereas a decrement in proximal muscles occurred in 64 percent [Stalberg and Sanders, 1981]. Similar findings have been described by other authors [Vial et al., 1991; Oh et al., 1992]. In a study of 27 juvenile myasthenic patients, Afifi and Bell found that the chance of finding a decrement doubled to 66 percent by including proximal muscles. In children with generalized disease at onset, 80 percent showed a decrement when proximal muscles were studied. In ocular MG, decrements are less common, occurring in 20–50 percent of patients [Stalberg and Sanders, 1981; Evoli et al., 1988]. Facial muscle RS should be included when clinical suspicion for anti-MuSK myasthenia exists, as facial muscles are much more prominently involved in this group [Muppidi and Wolfe, 2009]. RS at faster rates (i.e., 20 or 50 Hz) usually is not performed unless there is concern about LEMS, a rare condition in children.
Single-fiber electromyography
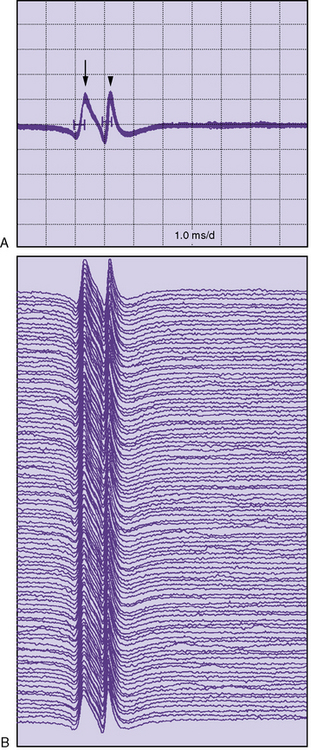
Single-fiber electromyography (EMG) is a more sensitive measure of neuromuscular transmission than RS and can be considered in select children. In MG, the time required for the EPP at the NMJ to reach threshold is extremely variable. The measurement of this variability in the EPP rise time is known as jitter. The jitter value, calculated in microseconds, is the most important piece of data obtained from single-fiber EMG. Everyone, including healthy individuals, has some degree of jitter (Figure 91-5). Myasthenic patients have increased jitter values (Figure 91-6). In addition, blocking occurs in myasthenics if a muscle fiber’s EPP never reaches threshold and depolarization does not occur. The frequency of blocking, expressed as a percentage, is also determined with single-fiber EMG (see Figure 91-6). In healthy individuals, the percentage of blocking is 0 percent.
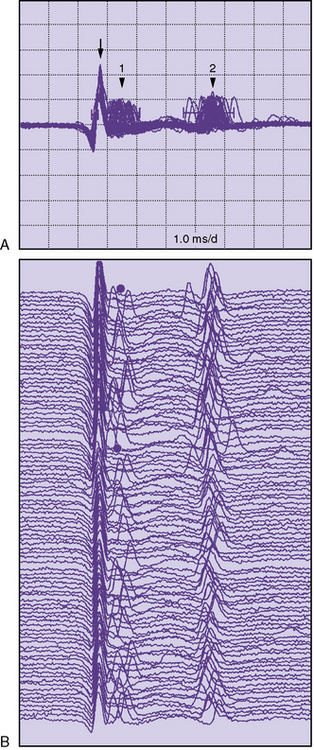
Single-fiber EMG is undoubtedly the most sensitive test for MG in adults. It is abnormal in 94 percent of generalized and 80 percent of ocular MG patients [Sanders and Howard, 1986; Oh et al., 1992]. However, single-fiber EMG has several disadvantages. It is a tedious and lengthy study that requires considerable patient cooperation and is poorly tolerated by many children. The need to use nondisposable single-fiber electrodes was also a limitation, but recently, normative data for single-fiber studies with disposable concentric needles have been published [Stalberg and Sanders, 2009]. Stimulated single-fiber EMG can be performed under sedation, requires less patient cooperation, and may be preferred in children, although it is still a lengthy procedure [Jabre et al., 1989]. An abnormal single-fiber EMG study is not specific for MG because increased jitter commonly occurs as a result of other neuromuscular diseases, including motor neuron disease, peripheral neuropathy, and many myopathies [Oh, 1988]. Fortunately, it is seldom necessary to perform single-fiber EMG to diagnose MG in children. It is probably most useful in children who present difficult diagnostic dilemmas and who otherwise have normal laboratory studies for MG. Single-fiber EMG abnormalities may be seen in 12–33 percent of first-degree relatives of patients with juvenile MG [Stalberg et al., 1976; Anlar et al., 1995]. Conventional needle EMG has limited diagnostic value in MG; however, short-duration, small-amplitude, early recruited myopathic units can be seen in anti-MuSK MG patients [Padua et al., 2006].
Antibody Testing
Anti-AChR antibodies
Anti-AChR antibody levels are not elevated in all MG patients. The assay is most helpful in adult generalized MG; it is positive in 85 percent of such patients [Lindstrom et al., 1976; Vincent and Newsom-Davis, 1985; Oh et al., 1992; Drachman, 1994]. Ocular MG patients, however, have measurable anti-AChR antibodies in only 50 percent of cases [Provenzano et al., 2009]. Children represent another group of MG patients who are often antibody-negative. In one study, 50 percent of prepubertal children with autoimmune juvenile MG were seropositive [Andrews et al., 1994]. Seropositive rates of 68 and 91 percent were observed in peripubertal and postpubertal disease onset, respectively. Similarly, seropositivity was more common in girls with onset of juvenile MG after 11 years of age [Anlar et al., 1996]. Seronegativity was more common in pure ocular forms, mild disease, and remission [Afifi and Bell, 1993].
Because congenital myasthenic syndromes and seronegative autoimmune MG present in early childhood, differentiating these disorders when the family history is negative is often difficult [Andrews, 2004]. Fluctuating weakness or disease severity and good responses to immunotherapy favor an autoimmune basis [Anlar et al., 1996].
The most common anti-AChR antibody test is the binding radioimmunoassay using bungarotoxin, measured in nanomoles per liter. The upper limit of normal varies among reference laboratories (usually between 0.03 and 0.5 nmol/L) [Lennon, 1982]. Other assays that block bungarotoxin binding to AChR (“blocking” assay) or that reduce the density of AChR on cultured human myotubes (modulating antibody assay) are also commercially available [Howard et al., 1987]. These additional assays may be useful in patients with suspected MG who test negative with the standard binding assay [Howard et al., 1987], but do not add significantly to the diagnostic sensitivity. Some laboratories offer all three (binding, blocking, and modulating) antibodies as one serological test. Recently, low-affinity anti-AChR antibodies against rapsyn-clustered AChR were seen in 66 percent of patients who were otherwise seronegative. These were mainly IgG1 antibodies that can activate complement C3b deposition [Vincent et al., 2008]. These new low-affinity AChR assays are not yet commercially available.
Anti-AChR antibody titers correlate poorly with MG severity [Roses et al., 1981]. Although the titer often falls as the clinical condition improves, antibody titers in general do not guide therapeutic decisions. Indeed, MG patients in clinical remission may still have elevated titers, but this is not an indication to continue immunosuppressive therapy.
Anti-MuSK antibodies
Since 2001, IgG from 40–70 percent of seronegative generalized patients has been found to bind to the extracellular domain of muscle-specific receptor tyrosine kinase (MuSK) [Hoch et al., 2001; Sanders et al., 2003; McConville et al., 2004]. Marked female predominance with mean age of onset in the fourth decade has been typical [Evoli et al., 2003; Sanders et al., 2003], although Evoli et al. encountered a child with disease onset at age 6 years, and two patients from the initial series presented before age 10 [Hoch et al., 2001]. The earliest reported onset of anti-MuSK MG is 2 years [Murai et al., 2006]. Three main patterns of anti-MuSK MG have been observed; one of them is clinically indistinguishable from anti-AChR generalized MG. The other two patterns are severe oculobulbar weakness and prominent neck, shoulder, and respiratory involvement, largely sparing ocular musculature. In these two phenotypic variants, limb strength is relatively intact [Sanders et al., 2003; Muppidi and Wolfe, 2009]. Anti-MuSK antibodies rarely occur in pure ocular MG [Wolfe et al., 2007].
It has been hypothesized that anti-MuSK antibodies impede agrin-mediated clustering of AChR and disrupt normal postsynaptic architecture [Jha et al., 2006]. We consider testing for anti-MuSK antibodies in all suspected MG patients who are anti-AChR antibody-negative. The assay is commercially available. Anti-MuSK MG is somewhat more refractory to conventional treatment when compared to anti-AChR MG [Pasnoor et al., 2009].
Striated muscle antibodies and other laboratory studies
Antibodies to striated muscle in MG patients were discovered before anti-AChR antibodies. These antibodies can be directed against a number of muscle proteins, including myosin, actin, alpha-actinin, titin, and ryanodine (RyR). It is generally believed that, if anti-striated muscle antibodies are present in an MG patient, they should raise suspicion for thymoma, as they are reported in up to 84 percent of patients with thymoma [Limburg et al., 1983]. However, these antibodies may be found in patients without thymoma and in patients with thymoma who do not have MG [Cikes et al., 1988; Romi et al., 2000]. The absence of anti-striated antibodies also does not rule out thymoma. From an adult perspective, anti-titin and anti-RyR antibodies have been forwarded as a marker for more severe disease in MG patients presenting after age 40 [Romi et al., 2000]. Thyroid function tests are routinely obtained at the time of initial evaluation, as thyroid disease often coexists with MG [Meriggioli and Sanders, 2009].
Treatment
Most patients with juvenile MG who require maintenance therapy are treated with anticholinesterase agents, with or without a variety of immunosuppressive medications. Pyridostigmine is recommended as an initial intervention [Snead et al., 1980; Saperstein and Barohn, 2004]. As with the adult form of the disease, corticosteroids and other immunomodulatory treatments are used, although few randomized controlled clinical trials have been performed in the pediatric MG population. Thymectomy plays an important role in treating older children at most centers. Plasmapheresis [Pinching and Peters, 1976] and intravenous gamma globulin [Arsura, 1989] are generally reserved for more refractory patients or for those in myasthenic crisis. Plasmapheresis and intravenous gamma globulin are also used to maximize function before thymectomy. Short-term supportive care and anticholinesterase agents are usually adequate for neonatal transient MG. Management for the different types of childhood MG is reviewed in the following sections.
Acetylcholinesterase Inhibitors
In juvenile MG, the aggressiveness of management should be in accordance with disease severity. In general, management attempts should first focus on pyridostigmine. A total daily dose of up to 7 mg/kg a day is delivered in 5–6 divided doses [Wolfe et al., 1997]. Older children can use 60 mg tablets that can be split in half as needed. Typical doses in older children and adults are 60 mg, 3–5 times a day. If symptoms are poorly controlled on a pyridostigmine dose exceeding 300 mg/day, it is probably necessary to add immunomodulating therapy. Sustained-release pyridostigmine is also available (Mestinon TS, 180 mg). However, because of variable absorption, difficulty adjusting doses, and increased side effects, many clinicians discourage use of this preparation.
Thymectomy
When a child’s symptoms can no longer be controlled by anticholinesterase agents alone, a decision must be made regarding whether to pursue thymectomy or immunosuppressive therapy. In 1939, Blalock and co-workers reported the remission of generalized MG in a 21-year-old woman after removal of the cystic remains of a necrotic thymic tumor [Blalock et al., 1939]. Since then, thymectomy, with or without the presence of thymoma, has gained widespread acceptance as a treatment for MG. Thus thymectomy was the first attempt at “immunotherapy” for MG and continues to be one of the most common treatments for the disease.
There is a general consensus that generalized MG patients between puberty and 60 years of age benefit from thymectomy [Rowland, 1987; Lanska, 1990]. However, randomized studies of thymectomy that control for medical therapy have never been performed. The use of thymectomy in very young children is controversial, in itself, because of concerns about a subsequent impairment in immune protection or an enhanced risk of cancer. A review of incidental thymectomy and thymectomy as treatment for MG in young children, however, did not find a consistent association between thymectomy and these proposed risks in children older than 1 year [Seybold, 1998]. In a survey that included 56 neurologists with MG expertise, lower age limits for performing thymectomy ranged from 1 year to puberty, with the median being 7.5 years [Lanska, 1990].
An evidence-based practice parameter from the American Academy of Neurology analyzed retrospective, controlled, nonrandomized studies of thymectomy in MG. A total of 28 studies published between 1953 and 1998 were identified [Gronseth and Barohn, 2000]. The effect of surgery was broadly favorable in most series. However, the benefit of surgery was generally small. For example, the median relative rate favoring surgery over nonsurgical treatment for achieving remission was 2.1 (a modest gain, considering that the median remission rate in the non-thymectomized groups was 10 percent). Other median relative rates were 1.6 for asymptomatic status, 1.7 for improvement, and 1.1 for survival. Patient subgroup analysis indicated that only those MG patients with moderate weakness or greater (Osserman 2b) showed a significant improvement following thymectomy compared to controls. Importantly, the modest benefits ascribed to thymectomy were confounded by baseline differences between the surgical and nonsurgical groups, as well as limited thymic resections performed in a significant percentage of the patients. No study included blinded assessments. As a result, the authors expressed uncertainty as to whether claims of improved MG outcomes were a result of thymectomy or related to differences in baseline characteristics between surgical and nonsurgical groups. The authors concluded that thymectomy should be considered a treatment option in patients without thymoma [Gronseth and Barohn, 2000]. To address this uncertainty, an international prospective, single-blinded, randomized trial in patients with non-thymomatous MG controlling for medical therapy is currently under way [Newsom-Davis et al., 2008]. In this study, patients are randomized to prednisone alone versus trans-sternal thymectomy with prednisone. The minimum age to enter into the study is 18 years.
Thymectomy has been widely used for the treatment of juvenile MG, showing a higher likelihood of remission when performed within 2 years of symptom onset compared to nonsurgical patients [Seybold, 1998]. In the largest study, 85 of 149 patients (57 percent) with juvenile MG underwent thymectomy. Of the thymectomized patients, 42 (49 percent) entered remission and another 25 (29 percent) improved clinically [Rodriguez et al., 1983]. In contrast, only 34 percent of patients who did not undergo thymectomy entered remission. Remission rates were 260 per 1000 person-years in the first year after thymectomy, falling to 95 per 1000 person-years over the next year after surgery [Rodriguez et al., 1983]. After 3 years, the remission rate decreased to 20 per 1000 person-years, similar to the spontaneous remission rate. Favorable predictors for postoperative remission included surgery during the first year after disease onset, bulbar symptoms, absence of ocular or generalized involvement, onset of symptoms between ages 12 and 16 years, and the presence of other autoimmune disorders [Rodriguez et al., 1983]. Gender of the child or thymic pathology did not appear to influence remission rates. Histologic findings of the thymus were normal in 16 percent, hyperplastic in 78 percent, and neoplastic in 3 percent.
Other series suggest that the clinical benefit from thymectomy may not be realized for as long as 7–10 years [Mulder et al., 1989]. These favorable clinical responses and the low morbidity and mortality of thymectomy in childhood cases bolster its widespread use [Batocchi et al., 1990], prompting some to consider the procedure at the onset of generalized symptoms [Adams et al., 1990]. Clinical improvement after thymectomy has been observed, whether or not there is a reduction in circulating anti-AChR antibody levels [Oosterhuis et al., 1985]. The role of thymectomy in patients with anti-MuSK antibody-positive myasthenia is unclear. Early reports suggested that most patients do not experience any benefit with thymectomy [Sanders et al., 2003]. Thymic tissue in the anti-MuSK antibody-positive population does not demonstrate morphologic abnormalities, and thymectomy does not produce significant clinical improvement for these patients [Lauriola et al., 2004]. Probability of achieving remission seems to be slightly higher in seronegative non-thymomatous patients when compared to anti-MuSK antibody-positive non-thymomatous patients [Ponseti et al., 2009].
The goal of thymectomy is to remove all of the thymic tissue. Trans-sternal thymectomy is the surgical technique used in the on-going thymectomy trial [Newsom-Davis et al., 2008]. Other approaches include transcervical, infra-axillary video-assisted thoracoscopic procedures, and various combinations of these with trans-sternal incisions [Jaretzki et al., 2004]. There is some evidence to support the view that the greater the resection, the better the long-term results [Jaretzki et al., 1988]. Using a “maximal” thymectomy approach that includes both transcervical and trans-sternal incisions, life-table analyses demonstrated an 81 percent remission rate at 7.5 years [Jaretzki and Wolff, 1988]. Comparative remission rates for transcervical approaches have been in the 30–45 percent range at 7 years [Jaretzki et al., 2003], and approximately 50 percent at 6 years using either an extended trans-sternal or a video-assisted thoracoscopic procedure that includes a transverse cervical incision [Mantegazza et al., 2003]. It should be noted that remission rates in surgical series often are unexpectedly high. Definitions of remission, as well as their duration, vary between studies, and the retrospective determination of these outcomes is certainly open to bias [Gronseth and Barohn, 2000]. Transcervical and video-assisted thoracoscopic procedures offer advantages from a cosmetic and postoperative recovery standpoint. Thoracoscopic procedures have been found to reduce the length of hospital stay and patient care costs when compared to open thoracic surgery [Hazelrigg et al., 1993].
The presence of a thymoma is, of course, the one absolute indication for thymectomy. All newly diagnosed MG patients must undergo computed tomography or magnetic resonance imaging of the chest to look for evidence of thymoma. Routine chest radiographs may not detect up to 25 percent of thymic tumors [Batra et al., 1987]. Although there is a consensus among neurologists that thymectomy should be considered in all but the youngest children with generalized MG who are not controlled on anticholinesterase agents alone, there is less enthusiasm for the procedure in pure ocular MG.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


