Chapter 22 Disorders of Neural Tube Development
Introduction
Neural tube defects (NTDs) are among the most frequently encountered congenital anomalies involving the central nervous system (CNS). Indeed, NTDs are second only to congenital heart defects as the most common serious birth defect, affecting between 0.5 and 10 per 1000 live births, depending on the population studied [Gelineau-van Waes and Finnell, 2001]. Rostral NTDs, such as anencephaly, that lead to an absent cranial vault and exposed brain are uniformly fatal. In contrast, most children born with posterior NTDs confined to the spinal column will survive with varying degrees of functional disability and shortened life span. NTDs may be classified in a number of ways but the most useful strategy is to assign the observed defect according to the suspected developmental abnormality. This allows a better appreciation of the severity of the CNS anomaly, which often correlates with the clinical picture and expected outcome. In this chapter, the term NTD will be used to encompass all defects that involve neural tube formation and differentiation. NTDs are also referred to as spinal or cranial dysraphism, depending on the level of the defect. Spina bifida formally refers to all types of spinal dysraphism, though it is often used synonymously with myelomeningocele (MMC), which is a severe and common form of spinal dysraphism in which meninges, along with spinal cord or axons, protrude outside of the vertebral column.
NTDs have been the subject of intensive research for decades and are considered to result from complex interactions of genes and environmental conditions. Indeed, over 200 gene defects in the mouse have now been associated with failed neural tube closure, and a number of these genes are likely to be involved in human NTDs as well [Harris and Juriloff, 2007; Gray and Ross, 2009; Harris, 2009]. This class of neurodevelopmental defect is particularly important to understand because a significant proportion of NTDs may be preventable with measures like prenatal maternal folic acid (FA) supplementation [MRC, 1991], by avoiding prenatal exposure to known teratogens like retinoids, and because medications often used in the treatment of epilepsy and neuropsychiatric disorders can increase the risk to pregnancies conceived while taking the drug [Ross, 2010]. This chapter will discuss the pathogenesis, risk factors, complications, and management of NTDs in children.
Anatomy and Embryology
Formation of the Neural Tube
Among the earliest morphological specializations in the embryo is the formation of the neural placode, followed by neural plate and then neural tube [Sadler, 2005]. At the end of the second gestational week, the human embryo is a bilaminar disc of epiblast cells overlying hypoblast cells [Sadler, 2004]. At the beginning of the third week, the disc develops a midline groove, the primitive streak, in the caudal third (Figure 22-1A), which marks the initiation of gastrulation and the formation of three germ layers – ectoderm (giving rise to skin and the nervous system), mesoderm (providing inductive signals to ectoderm and contributing to morphogenesis), and endoderm (giving rise to viscera). The primitive node at the cranial end of the streak contains cells that act to organize the embryonic axes (see Figure 22-1). At the same stage, thickening of the rostral ectoderm by the apical–basal elongation of cells into a pseudostratified columnar shape produces the neural placode that marks the initiation of neurulation (see Figure 22-1B) [Schoenwolf and Powers, 1987; Schoenwolf, 1988; Colas and Schoenwolf, 2001]. Cells migrating through the primitive streak and node displace the hypoblast cells to form endoderm and subsequently middle layer mesoderm. Cells that migrate through the node in the midline form the prechordal plate and notochord (Figure 22-2), which are important for induction of the ventral CNS structures, starting with the neural plate. The remainder of ectoderm surrounding the neural plate becomes epidermis.
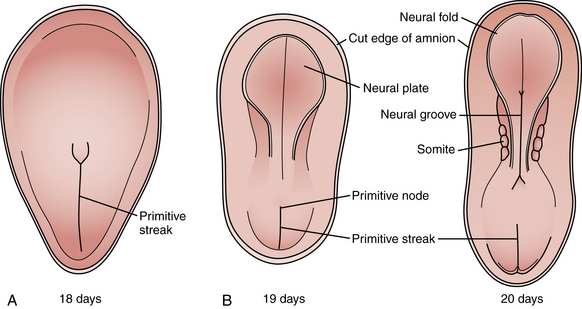
Fig. 22-1 Early stages of gastrulation and neurulation in human embryogenesis: views of the dorsal surface.
(Adapted from Sadler TW. Embryology of neural tube development. Am J Med Genet C Semin Med Genet 2005;135C:2–8.)
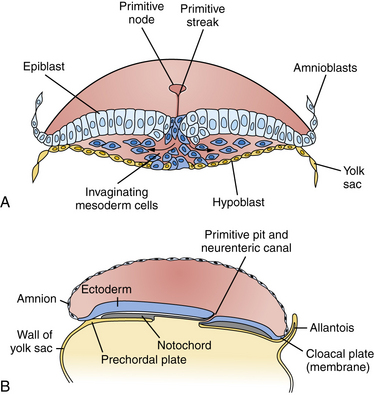
Fig. 22-2 Cross-sections through the primitive streak.
(Adapted from Sadler TW. Embryology of neural tube development. Am J Med Genet C Semin Med Genet 2005;135C:2–8.)
Neural plate formation is actually a default state of the ectoderm, and formation of epidermis involves the inhibition of bone morphogenetic protein (BMP) and the wingless (Wnt) signaling pathway [Harland, 2000]. As the plate begins to emerge, morphogenesis, or shape changes, involving groups of cells in the neuroepithelium and surround, is essential to formation of brain and spinal cord (Figure 22-3). By 20 days of gestation, the neural plate appears indented in the midline, forming a groove or medial hinge region flanked by ridges – the neural folds. These folds elevate from the plane of the neural plate through the combined influences of proliferation of neural cells and underlying mesenchymal cells [Greene and Copp, 2009]. Bending inward of the neural folds at the dorsolateral hinge points occurs through morphological shape changes of the neural cells, which become radially elongated, while their apical (luminal) poles constrict, through a combination of cell-cycle regulation that moves nuclei to the basal end of cells in the hinge region, and actin-myosin contraction at the apical poles of cells in the hinge region, to bring the tips of the neural folds into apposition [Greene and Copp, 2009]. In the head region, proliferation and movement of epidermis and mesenchyme cells also help to push the neural folds into apposition, while at spinal levels, neighboring mesenchyme may be less critical to neural tube closure [Greene and Copp, 2009]. In addition to cell elongation and proliferation, the shape changes in the neural plate are affected by cell motility in the form of convergent extension, in which laterally placed cells move to the midline and migrate rostrocaudally in a process that is mediated by noncanonical Wnt signaling (Figure 22-4) [Wallingford and Harland, 2001; Wallingford et al., 2002]. Thus, once elevation and bending of the neural folds occur, the lateral margins or tips of the folds join and then fuse in the midline to become the neural tube.
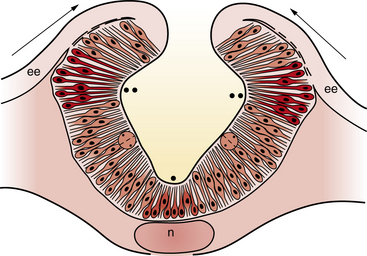
Fig. 22-3 A coronal cross-section through the cranial neural folds as they come together in the midline.
(Adapted from Sadler TW. Embryology of neural tube development. Am J Med Genet C Semin Med Genet 2005;135C:2–8.)
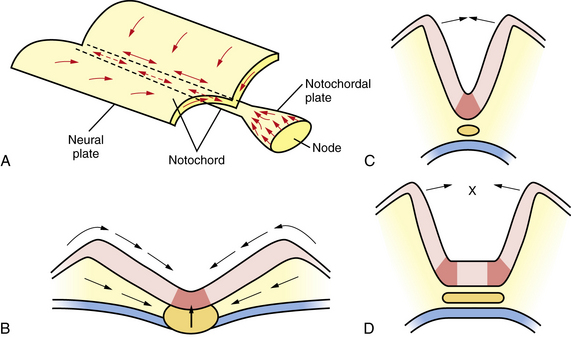
In order to achieve this rising and bending inward of the neural folds, the cells of the neural plate must proliferate in an ordered manner, called interkinetic nuclear migration of progenitors, forming a pseudostratified epithelium in which S-phase occurs at the basal (outer) surface of the neural folds, mitosis (M-phase) occurs at the apical (central or luminal) surface, and G1 and G2 phase nuclei are positioned at intermediate locations. In addition, in the process of convergent extension, cells move medially and through the medial hinge region to migrate rostrocaudally and elongate the neuraxis, narrowing the ventral floorplate (see Figure 22-3). If the floorplate is too wide or the neural folds fail to elevate and bend, the folds will not appose and NTDs will ensue (see Figure 22-4). The broadening of the ventral floorplate and shortening of the rostrocaudal axis reflect impaired convergent extension; they are demonstrated in the looptail mouse mutant, which bears a mutation in the Van Gogh-like 2 (Vangl2) gene that functions in the planar cell polarity (PCP) pathway [Ybot-Gonzalez et al., 2007]. When apposition is successful, midline fusion of the neural folds, a process known as neurulation, occurs first at primary closure points, and extends by adding multiple closure points like the teeth of a zipper rostrally and caudally from each node to complete neural tube closure [Pyrgaki et al., 2010]. In the mouse, there are typically three closure points:
Molecular Patterning of the Neural Tube
Work on frog, mouse, and zebrafish species has identified a host of factors that are necessary for proper neurulation and neural tube closure. These data have direct implications for human disease because vertebrates display a conserved body plan. A key observation was that a small fragment of non-neural tissue (mesoderm), when transplanted, was capable of duplicating the neural tube, indicating that secreted factors from mesoderm are sufficient to induce neurulation. Thus, the factors necessary for neurulation are intrinsic to and secreted from this mesodermal region. These factors include chordin, noggin, sonic hedgehog (SHH), and several others, which are both necessary and sufficient for proper neurulation and control of the amount and fate of the neuroectoderm (reviewed in De Robertis and Kuroda [2004]). In mammals, two distinct groups of non-neural cells appear to provide these early patterning signals: axial mesodermal cells of the notochord, which underlie the midline of the neural plate, and the cells of the epidermal ectoderm, which flank its lateral edges. The notochord is the source of ventralizing inductive factor, and the epidermal ectoderm is the source of dorsalizing factors. Opposing actions of these two signals appear to be critical in establishing the identity and pattern of cell types generated along the dorsal–ventral axis of the neural tube.
The nervous system is patterned along the anteroposterior, dorsal–ventral, and right–left axes in response to the expression of intrinsic patterning genes and critical embryonic signaling pathways involving secreted factors and cell–cell interactions [Altmann and Brivanlou, 2001]. The homeotic, Hox, transcription factor genes provide a positional “address” or identity of spinal cord and hindbrain neurons [Carpenter, 2002]. Fate determination along the dorsal–ventral axis also requires the action of three opposing signaling pathways:
In this model, SHH released by the notochord diffuses toward the ventral neural tube and induces the differentiation of the floor plate. The floor plate then becomes capable of producing additional SHH, which diffuses and establishes a gradient along the dorsal–ventral axis of the neural tube. Differentiation of cells in the dorsal half of the neural tube depends on signals such as BMPs provided by the lateral epidermal ectoderm. BMP4 and BMP7, released by the epidermal ectoderm, diffuse toward the dorsal neural tube and induce the differentiation of the roof plate. The roof plate then becomes capable of producing additional BMPs, which diffuse and establish a gradient along the ventral–dorsal axis of the neural tube. Regions of the spinal cord that are exposed to the highest concentrations of SHH and lowest concentrations of BMPs give rise to cells of ventral fate, including motor neurons, whereas cells exposed to lowest SHH and highest BMP concentrations give rise to cells of dorsal fate, such as commissural projection neurons [Cowan et al., 1997]. Fibroblast growth factor (FGF), NOTCH, and retinoic acid (RA) signaling is also known to act in dorsal–ventral patterning and neural fate determination, though their roles are less well understood. Rostrocaudally, the neural tube is regionalized into four major divisions: forebrain, midbrain, hindbrain, and spinal cord. Actions of FGF, Wnt and RA pathways confer a caudal identity [Diez del Corral and Storey, 2004; Aboitiz and Montiel, 2007] and also impact telencephalic patterning [Takahashi and Liu, 2006]. Homeotic Hox and Engrailed family transcription factors are particularly important for establishing segmental identities and morphogenetic features of hindbrain [Barrow et al., 2000; Cheng et al., 2010] (see also Chapter 24).
Epidemiology and Pathogenesis
Incidence
Among live births, females are more affected by NTDs than males by 2:1 [Elwood and Little, 1992; Little and Elwood, 1992; Shaw et al., 1994]. Additionally, the prevalence of NTDs varies across time, by region, and by ethnicity, and approximates 0.5 cases per 1000 in the United States but 10 per 1000 in parts of China and India [Northrup and Volcik, 2000; Gelineau-van Waes and Finnell, 2001]. These numbers do not reflect the decrease in incidence that has followed FA supplementation – with current estimates of 0.37 cases per 1000 reported in the US [Mills and Signore, 2004] – or the availability of prenatal diagnosis and the elective termination of some pregnancies with affected fetuses. The geographic and population variabilities in NTD rates are in keeping with the complexities of the etiology of neural tube closure failure, which involve both genetic and environmental factors [Janerich and Piper, 1978; Ross, 2010].
Over 30 years of clinical and basic science investigation are behind the insight that NTDs in humans are not caused by a single-gene defect but instead arise from the interplay of multiple genes, as well as gene–environment interactions [Holmes et al., 1976]. The twin concordance rate among same-sex twins (presumed monozygotic) is significantly increased (to 6.8 percent), supporting a genetic contribution [Janerich and Piper, 1978]. Furthermore, compared to an incidence in the general population of 1 per 1000, the risk of NTD recurrence in a family with one affected child increases to 1.8 per 100, but does not approach the 1 in 4 recurrence risk of an autosomal-recessive mutation with complete penetrance. Studies have documented the higher occurrence rate of NTDs in females compared to males, and the significantly higher incidence of consanguinity among parents of infants with NTDs [Elwood and Little, 1992; Shaw et al., 1994]. These and many other studies indicate prominent genetic, heritable contributions to failed neural tube closure that may well require the concerted action of multiple gene polymorphisms to manifest the disorder.
Complex Genetic Contributions
The complexity of the genetic underpinnings of neurulation is reflected in the NTD-prone mouse lines, for which over 200 mutations have been associated with failure of neural tube closure [Harris and Juriloff, 2007; Harris, 2009]. Despite this wealth of information, there is no single-gene polymorphism in the human homologs of these mouse genes that confers a robust, broadly reproducible enhanced risk of developing an NTD [Greene et al., 2009; Ross, 2010]. This has led to the supposition that either neurulation in the mouse is significantly different from that in humans, despite the similarities in morphogenesis and cell behaviors, or the genesis of NTDs requires the compounding of multiple gene polymorphisms. Supporting this latter premise, there are a number of examples in which modifier loci have been detected that increase the penetrance of NTD in mutant mouse models. A classic example is found the curly tail (ct) mouse, in which spina bifida is associated with a primary risk gene and at least three distinct modifier loci [Neumann et al., 1994]. The hypomorphic, partial loss-of-function mutation most responsible for NTDs in ct/ct mouse embryos occurs in a transcription factor called Grainy-head-like 3 (Grhl3). Another mutant with demonstrated multiple gene interactions is the SELHBc mouse, for which four loci have been mapped that conspire to result in exencephaly [Harris and Juriloff, 2007]. There are also examples of digenic mutations, or mutations in two genes, that must occur together to produce NTD in the mouse [Harris and Juriloff, 2007]. These digenic mutations in compound heterozygous mice affect processes from cell polarity (Cobl/Vangl2, Dvl1/Dvl2, Fzd3/Fzd6, Vangl2/Celsr1, Vangl2/Scrb, Vangl2/Ptk7), to actin cytoskeletal regulation (Enah/Vasp, Enah/Pfn1), to cell–cell adhesion (Gja1/Gja5, Itga1/Itga6), intracellular protein transport (Snx1/Snx2), intracellular signaling (Jnk1/Jnk2, Prkaca/Prkacb), or transcription regulation (Msx1/Msx2, Rara/Rarg). Genetic interactions that increase NTD risk certainly do occur in mice and can be expected to occur in humans as well [Ross, 2010].
The several hundred genes that have been associated with NTDs in mouse models are beginning to provide insights into molecular networks that are critically important for neurulation and may become clinically useful [Ross, in press]. For example, just as mutation in Vangl2 renders the looptail mouse prone to NTD, polymorphisms in human Vangl1 [Kibar et al., 2007] and Vangl2 [Lei et al., 2010] have recently been implicated in NTD patients. However, evidence that these single nucleotide polymorphisms (SNPs) can occur in unaffected individuals as well suggests that the picture is too incomplete at present to permit using these SNPs for risk assessment. The context of the SNP or genetic background and environmental factors must be taken into consideration. Toward establishing this capability, pathway relationships associated with NTDs are emerging that include PCP signaling pathways encompassing Wnt and SHH pathways, chromatin remodeling and DNA methylation pathways, cell-cycle regulation, apoptosis, and cytoskeletal regulation, as well as transcription factors [Harris and Juriloff, 2007; Harris, 2009; Ross, 2010]. The complexity of the genetic underpinnings of NTD indicates that meeting the challenge for determining individual risk – and the optimal preventative therapy – will require evaluation of multiple genes (in signaling, metabolic, and transcriptional pathways) in a single person to detect compounding effects of gene polymorphisms that alone might not be significant [Greene et al., 2009; Ross, 2010]. Advanced technologies for high-throughput genomic DNA sequencing and analysis, and detection of the epigenome and perhaps the microbiome, as well as untargeted metabolomic screening, will all play a role in the clinical evaluation of individual patient assessments of NTD risk and prevention.
Gene–Environment Interactions Influencing Neural Tube Defects
Environmental influences contributing to NTDs have long been recognized [Janerich and Piper, 1978; Zhu et al., 2009]. The variables that have been implicated as risk factors for nonsyndromic forms of spina bifida are listed in Table 22-1. Risk factors include maternal diabetes, in which both hyperglycemia and associated hyperinsulinemia increase NTD risk [Eriksson et al., 2003]. Maternal obesity and pre-pregnancy weight gain are also associated with increased NTD risk [Werler et al., 1996; Hendricks et al., 2001]. Moreover, maternal periconceptional elevations in simple sugars that raise the glycemic index have been associated with increased NTD risk, even among nondiabetic women [Shaw et al., 2003]. In animal models, exposing rat embryos to a hyperglycemic environment induces dysmorphisms, accompanied by increases in biomarkers of oxidative stress and inositol depletion [Wentzel et al., 2001].
Table 22-1 Risk Factors for Spina Bifida
| Risk Factor | Relative Risk (-Fold Increase) |
|---|---|
| Established Risk Factors | |
| History of previous affected pregnancy with same partner | 30 |
| Inadequate maternal intake of folic acid | 2–8 |
| Pregestational maternal diabetes | 2–10 |
| Valproic acid and carbamazepine | 10–20 |
| Suspected Risk Factors | |
| Maternal vitamin B12 status | 3 |
| Maternal obesity | 1.5–3.5 |
| Maternal hyperthermia | 2 |
| Maternal diarrhea | 3–4 |
| Gestational diabetes | NE |
| Fumonisins | NE |
| Paternal exposure to Agent Orange | NE |
| Chlorination disinfection byproducts in drinking water | NE |
| Electromagnetic fields | NE |
| Hazardous waste sites | NE |
| Pesticides | NE |
NE, not established.
(From Mitchell LE et al. Spina bifida. Lancet 2004;364:1885–1895.)
Gene–Diet Interactions in Neural Tube Defects: Role of Metabolism of Folic Acid and Other Nutrients
The important role of the folate pathway in the pathogenesis of NTDs has steadily gained acceptance since the landmark clinical investigations of Smithells and colleagues in the early 1980s prompted a large randomized trial of prenatal FA supplementation for women with previous NTD-affected pregnancies [Smithells et al., 1976; Smithells et al., 1983; MRC, 1991; Czeizel and Dudas, 1992]. Studies in the United Kingdom, later corroborated in Eastern Europe and elsewhere, indicated that the prevalence of NTDs could be reduced by 70 percent or more by prenatal supplementation with FA, even in the absence of maternal folate deficiency [MRC, 1991; Czeizel and Dudas, 1992]. However, some populations demonstrated only a small or no significant reduction in NTD rates with prenatal FA supplementation [Shaw et al., 1995], suggesting that differences in genetic background, diet, or other environmental exposures could impact the efficacy of folate supplementation. Inadequate intake of natural folate before and during early pregnancy is associated with a 2- to 8-fold increased risk of MMC and anencephaly, as indicated by several series of case-controlled, randomized clinical trials and community-based interventions [Wald, 1993]. Moreover, the risk of having a child affected by an NTD is indirectly related to both maternal folate intake and maternal folate intracellular level [Daly et al., 1995; Wald et al., 2001; Moore et al., 2003].
The mechanism underlying the association between NTDs and folate has not been established [Blom et al., 2006]. In view of the extraordinary variety of genes and molecular pathways contributing to NTD formation, perhaps the success of FA supplementation is due to the many biological functions to which folate metabolism contributes [Beaudin and Stover, 2009; Ross, 2010]. The exchange of single carbon units takes place in part in mitochondria, where serine cleavage generates glycine and formate, and glycine cleavage enzymes also generate formate that is transported to the cytoplasm. In the cytoplasm, formate enters the folate pathway for one-carbon metabolism (OCM) as formyl and then methenyl and methylene groups, added to or donated by dihydrofolates and tetrahydrofolates [Beaudin and Stover, 2007, 2009]. In the cytoplasm, the FA metabolic pathway is needed for physiological processes ranging from nucleotide biosynthesis, crucial for proliferation, to generation of pterin coactors impacting biochemical reactions, and generation of the body’s principal methyl donor, S-adenosyl methionine (SAM or Ado-met), which participates in methylation of DNA and histones modulating gene expression, and methylation of proteins and lipids [Blom et al., 2006; Beaudin and Stover, 2007; Miller, 2008].
Recent studies have demonstrated that NTDs can occur in the offspring of pregnant women who had maternal autoantibodies against folate receptors [Rothenberg et al., 2004]. These autoantibodies blocked the binding of 3HFA to folate receptors on placental membranes, presumably resulting in reduced levels of CNS folate that contributed to development of the NTD. This result was not replicated in a population study of a large Irish cohort [Molloy et al., 2009]. However, that research was weakened by the fact that a number of serum samples were taken from women up to 10 years after pregnancy. Another study, which was confined to measuring folate receptor antibodies in samples drawn during midgestation, found a significant association between antibody levels, the folate transport-blocking capability of the antibody, and NTD occurrence [Cabrera et al., 2008]. This may lead in future to clinical testing during first pregnancies that will be used to adjust the recommended level of FA supplementation in subsequent pregnancies.
Teratogens
RA is a well-known teratogen when administered to nonhuman embryos, in which one of its many effects is to induce NTDs, including spina bifida, exencephaly, and anencephaly in several different species [Shenefelt, 1972; Tibbles and Wiley, 1988; Yasuda et al., 1989]. Exposure to excess RA has also been implicated in human embryopathy [Lammer et al., 1985; Rosa et al., 1986]. In contrast, inactivation of RA-synthesizing enzyme genes or receptor genes in mice leads to significantly increased rates of NTDs. High doses of RA (vitamin A) or retinoids in medications such as isotretinoin (Accutane) are to be avoided during pregnancy, although vitamin A deficiency can also increase NTD risk [Azais-Braesco and Pascal, 2000].
Most, if not all, antiepileptic drugs (AEDs) are known teratogens [Ornoy, 2006]. Different AEDs, however, are associated with different constellations of malformations. An increased risk of MMC is associated with in utero exposure to valproic acid or carbamazepine alone, or in combination with other AEDs [Lammer et al., 1987; Dansky and Finnell, 1991; Matalon et al., 2002]. In infants exposed to valproic acid or carbamazepine, the risk of MMC can be as high as 1–2 percent [Koren and Kennedy, 1999; Zhu et al., 2009]. Women who use these drugs for indications other than epilepsy (e.g., bipolar disease, migraine, chronic pain) also are at increased risk of having a child with MMC if they become pregnant while taking these drugs. The mechanisms by which valproic acid and carbamazepine increase the risk of NTD have not been established, but there is general consensus that genetic predisposition to its teratogenic effect is required for valproate to promote NTDs. Among the pathogenic mechanisms associated with valproic acid are increased homologous recombination that induces mutations in the genome, generation of reactive oxygen species, and epigenetic reprogramming through direct inhibition of histone deacetylases by valproate (reviewed in Ross, 2010). Folate administration does not appear to protect against the effects of valproic acid or carbamazepine on neural tube closure.
Classification of Neural Tube Defects







Myelomeningocele
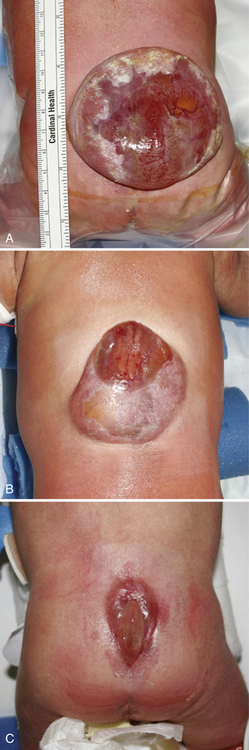
Myelomeningocele (MMC), the most complex of congenital spinal deformities, involves all tissue layers dorsal to and including the neural tube (i.e., spinal cord, nerve roots, meninges, vertebral bodies, skin). The dysplastic neural tube is a flat, disorganized segment of tissue located at the middle and most superficial portion of a cerebrospinal fluid-containing sac (Figure 22-5A, B). The surrounding epithelium may, in some cases, grow over the placode or, in some cases, the neural placode may remain in the spinal canal without a sac present. This condition is referred to as myeloschisis (Figure 22-5C), but probably is not substantially different from a typical MMC in embryologic terms. The absence of a sac probably occurs if cerebrospinal fluid flow in the spinal canal is obstructed, or if cerebrospinal fluid leaks from the spinal subarachnoid space into the amniotic cavity.
Antenatal Diagnosis
Maternal serum α-fetoprotein (AFP) determination and ultrasound examination are used to identify fetuses that have or are likely to have spina bifida or anencephaly [Drugan et al., 2001]. Positive findings from either of these two screening tests can be monitored with amniocentesis and detailed sonography [Kooper et al., 2007]. AFP is a component of fetal cerebrospinal fluid, and it may leak into the amniotic fluid from the open neural tube. Elevated amniotic AFP concentrations correlate with open NTDs, while closed lesions usually do not lead to increased AFP concentration. Detection of NTDs correlates with the magnitude of increase in the amniotic fluid AFP level; NTDs are associated in a minority of pregnancies with mildly elevated AFP levels, in a majority of those with moderately elevated levels, and overwhelmingly in those with very elevated AFP levels [Canick et al., 2003]. An AFP level of two times the normal median or higher is found in approximately 2 percent of unaffected pregnancies, 80 percent of open spina bifida pregnancies, and 95 percent of anencephalic pregnancies.
Sonography also can be used to differentiate between ventral wall defects and NTDs [Lennon and Gray, 1999], and to identify additional structural malformations that are characteristic of fetuses with chromosomal abnormalities [Sepulveda et al., 2004]. When a diagnosis of spina bifida is confirmed, ultrasound examination is used to assess spontaneous leg and foot motion and to screen for leg and spine deformities, a Chiari II malformation, and other physical defects. Ultrasonography can detect or confirm the extent of the NTDs [Watson et al., 1991] and has had an enormous impact on the number of liveborn infants among populations in which termination of pregnancy is accepted [Zlotogora et al., 2002]. It is 60 percent accurate in low-risk pregnancies, which is equivalent to the accuracy of serum AFP screening (64 percent), 89 percent accurate in high-risk pregnancies, and 100 percent accurate for women referred for confirmation of a suspected spina bifida by another ultrasonographer [Chan et al., 1993, 1995]. The data indicate that neither sonography nor AFP screening alone provides sufficient sensitivity or specificity, but that, when these studies are used together, the predictive value is much higher [Chan et al., 1995].
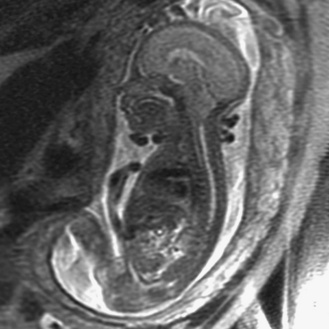
Prenatal magnetic resonance imaging (MRI), with ultra-fast T2-weighted sequences, also can be used to characterize the Chiari II and other malformations (Figure 22-6). Such prenatal imaging studies might help to predict neurologic deficit [Cochrane et al., 1996] and ambulatory potential [Biggio et al., 2001]. Most fetuses with spina bifida that are not electively terminated receive no treatment until after birth. Several studies have investigated whether method of delivery influences the outcome for infants with the disorder. A study based on a review of this work concluded that, in general, conclusive evidence is lacking that, relative to vaginal delivery, cesarean section improves the outcome in children with spina bifida [Anteby and Yagel, 2003]. Cesarean section, however, might be justified for large lesions, to reduce the risk of trauma, and is done after in utero treatment of spina bifida because the forces of labor are likely to produce a wound dehiscence.
Prevention
NTDs occur during early pregnancy, often before a woman knows she is pregnant; 50–70 percent of these defects can be prevented if a woman consumes sufficient FA daily before conception and throughout the first trimester of her pregnancy. Two landmark studies in the early 1990s found that administration of FA had a profound effect on the risk of NTDs [MRC, 1991; Czeizel and Dudas, 1992]. In 1992, to reduce the number of cases of MMC, the U.S. Public Health Service recommended that all women capable of becoming pregnant consume 400 μg of FA daily. Because approximately 50 percent of pregnancies in the United States are unplanned and the neural tube develops before most women know they are pregnant, it was also recommended that women consume this amount of FA routinely. Three approaches to increase FA consumption were cited:
Mandatory fortification of cereal grain products was legislated in the U.S. in 1996 and fully implemented as of January 1998. The estimated number of NTD-affected pregnancies in the U.S. declined from 4000 in the period 1995–1996 to 3000 in 1999–2000 [CDC, 2004; Mills and Signore, 2004]. This analysis controlled for declines in live births with NTDs as a result of recently implemented screening programs. Since the overall reduction of 25–30 percent in NTD rates in the U.S. since fortification was put into effect is below the expected 70 percent reduction, the debate continues regarding whether the fortification standard should be raised [Pitkin, 2007; Smith et al., 2008; Oakley, 2010].
Regardless of whether the optimal regimen for prenatal FA supplementation has been determined, there is still a significant proportion of pregnancies at risk for NTD for which FA will not effectively prevent NTD. Evidence in mouse models indicates that certain genetic backgrounds may include partial block in folate metabolism that can be circumvented by using alternative supplements. For example, the NTDs in the mouse mutant, Axial defects (Axd), are resistant to FA supplementation, but methionine – which will contribute to the methylation arm of the folate metabolic pathway – can reduce the occurrence of neurulation failure [Essien and Wannberg, 1993]. Another mouse mutant, curly tail (ct), which is prone to spina bifida that is not rescued by FA, can be protected against NTD by supplementation with myo-inositol [Greene and Copp, 1997]. Currently, clinical trials of prenatal inositol supplementation for the prevention of NTD are under way in the U.K. It appears likely that additional options for prevention of NTD will be available in the foreseeable future.
Clinical Features
The mortality rate for MMC is approximately 50 percent in the absence of therapy. Early surgery for closure of the lumbosacral defect is required to prevent meningitis. Later causes of death include hydrocephalus and renal failure. The renal complications are induced by chronic urinary tract infections, abnormal urodynamic function, and genitourinary tract abnormalities, such as progressive hydronephrosis. Patients at particular risk are the subgroup with high pressure within the bladder [Snodgrass and Adams, 2004].
MMCs may be situated at any longitudinal level of the neuroaxis. The location and extent of the defect determine the nature and degree of neurologic impairment; rating scales have been developed in an attempt to standardize the evaluation of affected children [Oi and Matsumoto, 1992]. Lumbosacral involvement is most common. Thoracic defects are the most complex and frequently are associated with serious complications. Cervical cord involvement is different from MMC of the lower spine and can be differentiated into two types:
Cervical lesions are clearly protuberant, covered at the base by full-thickness skin, and covered on the dome by a thick epithelium. Neural tissue is not superficially exposed but usually is tethered to adjacent dural or intrasaccular tissues. These differences likely are responsible for the more favorable outcome in cervical lesions [Meyer-Heim et al., 2003]. Varying degrees of paresis of the legs, usually profound, and sphincter dysfunction are the major clinical manifestations. Congenital dislocation of the hips or deformities of the feet, such as clubbing, may also occur. Severe sensory loss and accompanying trophic ulcers may complicate the condition. Occasionally, only sphincter disturbances are present. Radiographs reveal the primary defect of the vertebral arch.
Hydrocephalus is present in about 70–85 percent of patients with MMC, and occurs most frequently when the lesion is situated in the thoracolumbar area [Rintoul et al., 2002], which is the case in 90 percent of patients. Most persons with spina bifida have normal intelligence, but specific cognitive disabilities and language difficulties are common and can adversely affect educational and occupational achievements and the ability to live independently [Northrup and Volcik, 2000; Vachha and Adams, 2003].
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


