Chapter 74 Disorders of Upper and Lower Motor Neurons
Disorders of Upper Motor Neurons
Neuroanatomy of Upper Motor Neurons
The UMN is a motor neuron, the cell body of which lies within the motor cortex of the cerebrum, and the axon of which forms the corticobulbar and corticospinal tracts. The LMNs, lying in the brainstem motor nuclei and the anterior horns of the spinal cord, directly innervate skeletal muscles. The UMNs are rostral to the LMNs and exert direct or indirect supranuclear control over the LMNs (Box 74.1).
Box 74.1 Upper Motor Neurons and Their Descending Tracts
Motor Cortex
Limbic Motor Control
The limbic system is involved in emotional experience and expression and associated with a variety of autonomic, visceral, and endocrine functions. It strongly influences the somatic motor neurons. The emotional status and experience of an individual determines overall spinal cord activity, and the limbic motor system also influences respiration, vomiting, swallowing, chewing, and licking (at least in animal studies). Furthermore, the generation of signs of pseudobulbar hyperemotionality (pseudobulbar affect, emotional incontinence) in ALS is closely related to an abnormal limbic motor control, particularly in the periaqueductal gray and nucleus retroambiguus. The latter nuclei project to the somatic motor neurons that innervate pharyngeal, soft palatal, intercostal, diaphragmatic, abdominal, and probably laryngeal muscles. Pseudobulbar hyperemotionality symptoms may appear when UMN control over these motor nuclei is impaired, and thus limbic motor control is disinhibited. There appears to be some degree of emotional regulation by the cerebellum. The “cerebellar cognitive affective syndrome” can arise when stroke, tumor, or infection interrupts connections between the cerebellum and cerebral association and paralimbic regions (Schmahmann and Sherman, 1998).
Signs and Symptoms of Upper Motor Neuron Involvement
Loss of Dexterity
Loss of dexterity is one of the most characteristic signs of UMN impairment. Voluntary skillful movements require the integrated activation of many interneuron circuits in the spinal cord; such integration is ultimately controlled by the corticospinal tract and thus by UMNs. Loss of dexterity may express itself as stiffness, slowness, and clumsiness in performing any skillful motor actions. Asking the patient to perform rapid repetitive motions such as foot or finger tapping assesses loss of dexterity at the bedside. It is useful to assess both sides of the body, as many motor neuron disorders are asymmetrical (Box 74.2).
Box 74.2 Signs and Symptoms of Upper Motor Neuron Involvement
Loss of muscle strength (mild weakness)
Pathological reflexes (Babinski, Hoffmann sign, loss of abdominal reflexes)
Increased reflexes in an atrophic limb (probable upper motor neuron sign)
Pseudobulbar (spastic bulbar) palsy (emotional lability, brisk jaw jerk, hyperactive gag, forced yawning, snout reflex, suck reflex, slow tongue movements, spastic dysarthria)
Laboratory Evidence of Upper Motor Neuron Involvement
Neuroimaging
The use of brain magnetic resonance imaging (MRI) in ALS is largely to exclude other conditions but sometimes shows abnormal signal intensity in the corticospinal and corticobulbar tracts as they descend from the motor strip via the internal capsules to the cerebral peduncles. In ALS, signal changes, best appreciated on proton density images of the internal capsules, probably represent wallerian degeneration; similar changes also appear on conventional T2-weighted and fluid-attenuated inversion recovery (FLAIR) sequences. However, these changes do not appear to be sufficiently sensitive, and efforts continue to evaluate other potential MRI techniques such as diffusion tensor and volumetric MRI, which may serve as markers of UMN disease (Agosta et al., 2010).
Magnetic Resonance Spectroscopy Imaging
Proton density magnetic resonance spectroscopy (1H-MRS) is a noninvasive nuclear magnetic resonance technique that combines the advantages of MRI with in vivo biochemical information. A significant reduction of N-acetylaspartate, a neuronal marker, relative to creatine or choline (used as internal standards) exists in the sensorimotor cortices of patients with ALS who have UMN signs. Alterations in the measured levels of these metabolites using 1H-MRS are useful in the detection of UMN dysfunction early in the evolution of ALS and are useful to monitor progression over time. MRS still requires further technological improvements before it comes into widespread use (Mitsumoto et al., 2007).
Primary Lateral Sclerosis
Primary lateral sclerosis (PLS), first described by Erb in 1875, is a rare UMN disease variant that accounts for 2% to 4% of all cases of ALS and is traditionally distinguished by a lack of LMN involvement. The Pringle criteria for PLS stipulated that disease be restricted to the UMN system for at least 3 years from the time of clinical onset (Pringle et al., 1992), but a figure of 4 years is now proposed during which there is neither clinical nor neurophysiological evidence of LMN involvement. In a recent study comparing the evolution of disease in PLS versus UMN-predominant ALS and typical ALS, the median time to development of electromyographic (EMG) LMN features after onset in those with an evolving ALS was 3.17 years; in those patients, clinical signs of LMN disease occurred on average about 6 months later. Nonetheless, later development of LMN signs may occur and require reclassification as ALS in some cases, which therefore necessitates constant longitudinal review of each case (Gordon et al., 2009).
PLS typically presents in patients in their early 50s (about a decade younger than typical MND/ALS patients) as a very slowly evolving spastic paraparesis that spreads to the upper limbs and eventually causes pseudobulbar palsy. In rare instances, onset is in the bulbar system or follows a slowly ascending or descending hemiplegic pattern (Mills hemiplegic variant), but a bulbar-onset presentation should make the clinician wary of later LMN signs elsewhere. Other features include cramps and fasciculations, but such complaints are neither prominent nor universal. Bladder dysfunction is rare and, if it occurs at all, tends to be a late feature. Although muscle weakness is present, the main deficits are due to spasticity in dexterity and gait. The rate of progression can be exceedingly slow, often progressing over many years to the point where the patient manifests a robotic gait, debilitating generalized spasticity, and prominent pseudobulbar palsy. Muscle atrophy, if it occurs at all, is a very late feature. No clinically detectable sensory changes occur. Neuropsychological test batteries may define subtle cognitive deficits due to frontal cortical involvement, but dementia is not a prominent feature. A few patients may exhibit abnormal voluntary eye movements. Breathing is usually unimpaired in PLS, and as a consequence, forced vital capacity (FVC) is not affected (Gordon et al., 2009).
The prognosis is significantly better than for MND/ALS: one series had a median disease duration of 19 years and another series exhibited a range of survival from 72 to 491 months (Murray, B., 2006). The underlying pathogenesis of PLS remains undefined. Pathological changes include a striking loss of Betz cells in layer 5 of the frontal and prefrontal motor cortex (and other smaller pyramidal cells) together with laminar gliosis of layers 3 and 5 and degeneration of the corticospinal tracts. Spinal anterior horn cells are characteristically unaffected.
Diagnosis
The diagnosis of PLS is essentially one of exclusion (Table 74.1). Rare reports exist of UMN-onset ALS exist where the interval between onset of UMN signs and subsequent LMN signs have been up to 27 years. As such, it is vital to reassess patients diagnosed with PLS, as late signs of LMN involvement may occur that would reclassify their disorder as UMN-onset ALS.
Table 74.1 Disorders of Upper Motor Neurons and Their Key Characteristics
| Disorders | Key Characteristics |
|---|---|
| Primary lateral sclerosis | A diagnosis of exclusion |
| Hereditary spastic paraplegia | Heredity, usually autosomal dominant, spastin gene mutation, other mutations (see text), “sporadic” |
| HTLV-1-associated myelopathy | Slowly progressive myelopathy, endemic, and positive HTLV-1 |
| HTLV-2-associated myelopathy | Amerindian, IV drug abuser, concomitant HIV |
| Adrenomyeloneuropathy | X-linked recessive inheritance, adrenal dysfunction, myelopathy, very long-chain fatty acid assay |
| Lathyrism | History of consumption of chickling peas |
| Konzo | Eastern African, cassava root consumption |
HIV, Human immunodeficiency virus; HTLV, human T-lymphotropic virus; IV, intravenous.
Appropriate testing must exclude all definable causes for generalized UMN involvement. These include structural abnormalities (Chiari malformation and intrinsic and extrinsic spinal cord lesions) and myelopathies such as multiple sclerosis (MS) spondylotic cervical myelopathy, human immunodeficiency virus (HIV) myelopathy, human T-lymphotropic virus type 1 (HTLV-1) myelopathy, Lyme disease, syphilis, or adrenomyeloneuropathy. Spondylotic cervical myelopathy and MS are probably the most common causes among these disorders. The family history must be negative to rule out hereditary spastic paraplegia (HSP)/familial spastic paraparesis, spinocerebellar ataxia (SCA), hexosaminidase-A (Hex-A) deficiency, familial ALS (FALS), or adrenomyeloneuropathy. It is now apparent that some spastin mutation–associated HSP may lack a family history; it is worthwhile to carry out this gene test in patients presenting with symptoms and signs that are restricted to the lower extremities (Brugman et al., 2009). Paraneoplastic syndromes (especially in association with breast cancer) and Sjögren syndrome may clinically resemble PLS. Understanding of these entities is poor.
Treatment
No specific pharmacotherapy is available, and treatment therefore focuses on symptom control and supportive care. However, antispasticity drugs such as the GABA-B agonist, baclofen, and the central α2-agonist, tizanidine, may be tried for symptomatic treatment. Severe spasticity sometimes requires the insertion of an intrathecal baclofen pump. Tricyclic antidepressants, selective serotonin reuptake inhibitors, or dextromethorphan/quinidine may control pseudobulbar affect lability (Brooks et al., 2005).
Hereditary Spastic Paraplegia
HSP (or familial spastic paraparesis) is a genetically and clinically heterogeneous group of disorders rather than a single entity. The clinical feature common to all cases is progressively worsening spasticity of the lower extremities, often with variable degrees of weakness. The characteristic pathology is retrograde degeneration of the longest nerve fibers in the corticospinal tracts and posterior columns. Its estimated prevalence is 0.5 to 11.9 in 100,000, but its worldwide prevalence may actually be underestimated because of the benign nature of the disease in many families. Although the most common mode of inheritance is autosomal dominant, it may also be inherited in a recessive or X-linked fashion, and 12% to 13% of cases with apparently sporadic spastic paraparesis have spastin mutations (Depienne et al., 2006).
Genetic linkage studies of families around the world have mapped loci to over 40 autosomes as well as the X chromosome, with 17 distinct genes identified to date (Salinas et al., 2008). Inheritance of most pure HSP is autosomal dominant, whereas complicated forms are more often autosomal recessive. Between 40% and 45% of all families link to the SPAST gene on chromosome 2p22-21, which encodes spastin, a 616-amino acid protein. Mutations of various types (missense, nonsense, frameshift, splice site) may affect this gene (McDermott, C.J., 2006). Spastin is a highly conserved member of the AAA family of proteins (adenosine triphosphatase [ATPase] associated with various cellular activities). The exact role of mutant spastin in the pathogenesis of HSP is undefined, although a disturbance in maintenance of the microtubule cytoskeleton may exists, thus disrupting axonal transport. More than half of all cases do not manifest symptoms and signs until after age 30 years. Although this is normally a pure HSP, complicated forms occur, and some cases can develop a late-onset cognitive decline. Pathologically, degeneration of the longest corticospinal tracts and, to a lesser degree, the posterior columns of the spinal cord is seen.
Mutations in the SPG3A gene on 14q11-q21 encoding the novel protein, atlastin, give rise to an autosomal dominant often early-onset (<10 years of age) pure HSP which accounts for about 10% of autosomal dominant cases. This protein shares structural homology to guanylate-binding protein 1, which is a member of the dynamin family. Dynamins are important in intracellular trafficking of various kinds of vesicles. Mutations in KIF5A (SPG10, Chr 12q), a kinesin motor domain that is critical in intracellular transport, can cause both early- and late-onset spastic paraparesis with distal amyotrophy (Blair et al., 2006). Spastic paraplegia 11 (SPG11) is an autosomal recessive complicated HSP (thin corpus callosum, neuropathy, cognitive impairment) due to mutations in the spatacsin gene on chromosome 15q. This protein is of unknown function and does not appear to interact with the Golgi apparatus or microtubules. The cause of autosomal dominant pure HSP, linked to 2q24-34, is a mutation in the SPG13 gene, which encodes a mitochondrial heat shock protein. Recessively inherited complicated HSP links to chromosome 16q and is caused by a mutation in a gene encoding a mitochondrial protein known as paraplegin; this disorder can be ether pure or complicated (cerebellar signs, pale optic discs, and peripheral neuropathy). The genes for two different X-linked complicated HSP have been identified. In the first, mutant L1 (neural) cell adhesion molecule (L1CAM) may disrupt neuronal migration or differentiation; in the second mutant proteolipid protein (PLP1) is found in association with changes in white matter (duplication mutations in this same gene can also cause Pelizaeus-Merzbacher disease). Spastic paraplegia 17 (SPG17) is caused by mutations in the seipin gene on chromosome 11q12-q14. Also known as Silver syndrome, this disorder is an autosomal dominant complicated form of HSP with distal hand and foot amyotrophy beginning in the late teens to early 30s. Mutations in this gene are also the cause of a form of distal hereditary neuropathy (Charcot-Marie-Tooth [CMT] disease type 5).
Diagnosis
The basis for diagnosis of HSP is evidence of a family history in the setting of progressive gait disturbance, evidence of lower-extremity spasticity, and sparing of craniobulbar function. However, difficulties arise when there is no clear family history in recessive or X-linked disease and in cases of sporadic spastin mutation–associated HSP. Furthermore, considerable variation in disease expression exists between and within HSP families. In the absence of a family history or a demonstration of a known mutation, it is important to consider alternative causes for the clinical presentation, including structural disease (e.g., cerebral palsy, hydrocephalus, myelopathy), degenerative/infiltrative/inflammatory disease (e.g., MS, ALS, SCA, leukodystrophy), infections (syphilis, HIV, HTLV), levodopa-responsive dystonia, metabolic/toxic damage (vitamin B12 deficiency [subacute combined degeneration of the spinal cord (SCDC)], vitamin E deficiency, copper deficiency, lathyrism), and paraneoplastic disorders. MRI may reveal that cervical and thoracic spinal cord diameters are significantly smaller in both pure and complicated HSP than in controls (Sperfeld et al., 2005). Perhaps the most important differential diagnosis is that between apparently sporadic pure HSP and PLS, especially as the later may present with a slowly evolving spastic paraparesis for many years prior to the development of upper limb or bulbar features. The only reliable way to distinguish such disorders is through genetic testing; age at onset, urgency of micturition, and signs of dorsal column involvement (clinical or abnormal somatosensory evoked potentials [SSEPs]) are not accurate indicators of HSP versus PLS (Brugman et al., 2009).
Human T-Lymphotropic Virus Type 1–Associated Myelopathy, or Tropical Spastic Paraparesis
HTLV-1 causes a chronic progressive myelopathy that is referred to as tropical spastic paraparesis (TSP) in the Caribbean or HTLV-1–associated myelopathy (HAM) in Japan. This retrovirus is endemic in the Caribbean area, southwestern Japan, equatorial Africa, South Africa, parts of Asia, Central America, and South America, where it infects between 10 and 20 million people. Transmission occurs though sexual contact, intravenous (IV) drug use and also through breastfeeding. While between 2% and 3% of those infected can develop adult-onset T-cell leukemia, an estimated 2.5% to 3.8% can develop a chronic inflammatory myelopathy, with up to 20/100,000 affected in the Caribbean population and 3/100,000 in Japan. Recent evidence implicates high levels of activated HTLV-1–specific helper T cells and cytotoxic T cells in the pathogenesis of this syndrome; these immune cells appear to activate in response to interactions with retroviral env and tax proteins with greatest activity within the thoracic cord. Increased susceptibility for neurological disease appears to depend on both viral and host factors, with differences in certain HTLV-1 subgroups, proviral load, and HLA background being important. This may also explain differences in susceptibility between ethnic populations (Saito, 2010). Mode of transmission is through contaminated blood, sexual activity, breastfeeding, and very rarely in utero.
HAM/TSP is a chronic, insidiously progressive myelopathy that typically begins after age 30 years (but can occur as early as the first decade). In addition to slowly progressive spastic paraparesis, patients complain of lower-extremity paresthesias, a painful sensory neuropathy, and bladder dysfunction, and some patients may also develop optic neuropathy. Examination reveals UMN signs in the legs (weakness, spasticity, pathological reflexes, hyperreflexia), although reflexes may also be brisk in the arms. Overall, evidence of LMN involvement may be scant, and objective sensory findings may be difficult to detect. MRI may reveal increased signal on T2-weighted sequences in periventricular white matter and atrophy of the thoracic cord, but these findings may not be specific to HTLV-1. The definitive diagnosis of HAM/TSP requires HTLV-1–positive serology in blood and cerebrospinal fluid (CSF). To be sensitive and specific, CSF should reveal a combination of a polymerase chain reaction amplification of HTLV-1 deoxyribonucleic acid (DNA), together with evidence of an increased HTLV-1–specific antibody index and oligoclonal bands (Puccioni-Sohler et al., 2001). At present, no antiviral agents effectively treat HAM/TSP, but a case report showed partial benefit of plasmapheresis (Narakawa et al., 2001). As more is learned about the molecular etiology of HAM/TSP, future therapies will likely target the pathogenic effect of HTLV-1–reactive T cells.
Human T-Lymphotropic Virus Type 2–Associated Myelopathy
Though phylogenetically similar in many respects, HTLV-1 and HTLV-2 are still antigenically distinct. Nonetheless, using enzyme-linked immunosorbent assay (ELISA) and Western blot techniques, many laboratories worldwide often report the presence of sero-indeterminate HTLV-1/2. It has long been thought that myelopathy in such sero-indeterminate cases is due to HTLV-1 rather than HTLV-2, but rare cases are now being described of a syndrome characterized by spastic paraparesis, diffuse hyperreflexia, spastic bladder, and periventricular white matter changes on MRI in patients infected with HTLV-2 but not HTLV-1. This retrovirus is endemic in some Native American tribes and now often encountered worldwide among IV drug abusers. It is worthwhile to test CSF and serum for the presence of this virus in known IV drug abusers who present with a spastic paraparesis (Silva et al., 2002). However, co-infection with HIV-1 is a confounding factor in many cases of presumed HTLV-2–associated neurological disease. It has been suggested that such co-infection, rather than infection with HTLV-2 alone, increases the likelihood of neurological manifestations (Araujo and Hall, 2004; Posada-Vergara et al., 2006).
Adrenomyeloneuropathy
Adrenomyeloneuropathy is a variant of adrenoleukodystrophy, an X-linked recessive disorder caused by mutations in the ABCD1 gene on chromosome Xq28 that encodes a ubiquitously expressed integral membrane peroxisomal ATPase-binding cassette transporter protein. Mutations in this gene lead to abnormal peroxisomal β-oxidation, which results in the harmful accumulation of very long-chain fatty acids (VLCFAs) in affected cells. Excessive levels of VLCFAs may interfere with the membrane components of both neurons and axons. The most common phenotype, adrenoleukodystrophy, is an inflammatory disorder of brain and spinal cord that affects young boys 4 to 8 years of age, who develop severe adrenal insufficiency, progressive cognitive deterioration, seizures, blindness, deafness, and spastic quadriparesis. Adrenomyeloneuropathy is a noninflammatory axonopathy of the spinal cord that involves descending corticospinal tracts in the thoracic and lumbosacral regions and the ascending posterior columns in the cervical region. The characteristic clinical picture is a slowly progressive spastic paraparesis and mild polyneuropathy in adult men (in their late 20s), with or without sensory symptoms and sphincter disturbances. Adrenal insufficiency may be present and may predate onset of neurological symptoms by several years. Adult female carriers may present with slowly progressive spastic paraparesis. Approximately 20% of men with adrenomyeloneuropathy also develop cerebral changes on MRI that may accompany cognitive/language/behavioral deterioration. Rare cases may present as a spinocerebellar degeneration. Considerable phenotypic variation exists even within individual families. Female carriers may manifest more subtle symptoms such as cramps, back pain, or arthralgias. The diagnosis should be suspected in male cases with progressive sensorimotor deficits in the legs and a family history of a myelopathy (including supposed MS). Progressive sensorimotor deficits in the lower extremities with a history of memory loss or “attention deficit disorder” should also prompt testing for adrenomyeloneuropathy, as should a history of idiopathic childhood epilepsy or primary adrenal failure (Mukherjee et al., 2006). Sural nerve biopsies show loss of both myelinated and unmyelinated axons, with some degree of onion bulb formation. Ultrastructural examination may show characteristic inclusions (empty lipid clefts) in Schwann cell cytoplasm. Nerve conduction studies and needle electrode examination may reveal a predominantly axon-loss type of sensorimotor polyneuropathy with a lesser component of demyelination, and SSEPs may show reduced or absent responses. The diagnostic test of choice is to demonstrate increased VLCFA levels in plasma, red blood cells, or cultured skin fibroblasts. No specific therapy exists for adult-onset adrenomyeloneuropathy.
Plant Excitotoxins
Lathyrism
Lathyrism is a chronic toxic nutritional neurological disease caused by long-term (or subacute) ingestion of flour made from the drought-resistant chickling pea (Lathyrus sativus). It is an important example of a disease in which a natural excitotoxin causes selective UMN impairment. The responsible neurotoxin is β-N-oxalylamino-l-alanine (BOAA), an α-amino-3-hydroxyl-5-methyl-4-isoxazolepropionic acid (AMPA) glutamate receptor agonist. Ingestion of this neurotoxin results in increased intracellular levels of reactive oxygen species and subsequent impairment of the mitochondrial oxidative phosphorylation chain. Degeneration is most prominent in those Betz cells of the motor cortex (and the longest corresponding pyramidal tracts) that subserve lower-extremity function. Lathyrism occurs in the indigenous populations of Bangladesh, China, Ethiopia, India, Romania, and Spain. It also occurred in regional concentration camps during World War II. The condition may occur in epidemic form when malnourished populations increase consumption of flour made from L. sativus chickling peas during times of food shortage due to droughts. An analysis of an epidemic of neurolathyrism in Ethiopia showed a higher incidence in boys aged 10 to 14 years. The increased risk was associated with cooking grass pea foods in traditional clay pots (Getahun et al., 2002). The onset of clinical toxicity is either acute or chronic, manifesting as muscle spasms, cramps, and leg weakness. In addition to spastic paraparesis, sensory (including leg formications) and bladder dysfunction may occur. Occasionally there is a coarse tremor of the upper extremities. Although irreversible, the disorder is not progressive (unless there is continuing intoxication), and lifespan is not affected.
Disorders of Lower Motor Neurons
Clinical Features of Lower Motor Neuron Involvement
Loss of Muscle Strength (Weakness)
In a disease causing chronic motor unit depletion, neighboring axons belonging to healthy motor neurons may reinnervate denervated muscle fibers belonging to a diseased motor unit by collateral sprouting. In this way, existing motor units continually modify in the face of persistent losses of motor axons to maintain muscle strength. For example, in patients who have recovered from acute poliomyelitis, depletion of more than 50% of LMNs occurs before residual muscle weakness is clinically detectable. Healthy individuals have sufficient motor units available to offset an unexpected loss of motor neurons (Box 74.3).
Fasciculations
Fasciculations are spontaneous contractions of muscle fibers belonging to a single (or part of a) motor unit. (A video of this disorder can be found at www.expertconsult.com) Clinically, fasciculations appear on the muscle surface as fine, rapid, flickering, and sometimes vermicular contractions that occur irregularly in time and location. The impulse for the fasciculation appears to arise from hyperexcitable motor axons anywhere in their course. Fasciculations can occur both in healthy individuals and in patients with LMN involvement, so fasciculations themselves do not indicate LMN disease.
 (see video). Large fasciculations may occur in muscles undergoing extensive chronic reinnervation, such as chronic spinal muscular atrophy (SMA), Kennedy disease, and the postpoliomyelitis syndrome.
(see video). Large fasciculations may occur in muscles undergoing extensive chronic reinnervation, such as chronic spinal muscular atrophy (SMA), Kennedy disease, and the postpoliomyelitis syndrome.Laboratory Evidence of Lower Motor Neuron Involvement
Electrodiagnostic Examination
The electrodiagnostic examination (EDX) consists of nerve conduction studies and needle electrode examination (see Chapter 32B). The loss of motor units reflects in the loss of the amplitude of the maximal compound muscle action potential (CMAP). In a primarily demyelinating process, conduction velocity slows, and in severe cases, block. In a primarily axon loss process, there is usually only a modest degree of conduction velocity slowing commensurate with dropout of large myelinated axons. Sensory nerve conduction studies are normal in pure LMN disorders.
Acute Poliomyelitis
Clinical Features
After a brief 3- to 6-day incubation period, a viremia occurs, during which approximately 90% of individuals remain asymptomatic. Most of the remaining individuals develop an acute flulike illness with cough, malaise, diarrhea, myalgia, headache, and fever. This self-limited “abortive” polio usually lasts 2 to 3 days, and patients do not progress to develop acute muscle weakness. Between 2% and 3% of acutely infected patients develop aseptic meningitis characterized by severe headache due to meningeal irritation. This is typically self-limited and resolves within 7 to 14 days. Less than 1% of infected patients who ingest poliovirus develop the acute paralytic syndrome, characterized by localized fasciculations, severe myalgia, hyperesthesias, usually fulminant focal and asymmetrical paralysis, and fever. Any skeletal muscle can weaken, including bulbar muscles and muscles of respiration, but the leg muscles are the most commonly affected (Howard, 2005).
Laboratory Features
Motor nerve conduction studies performed 21 or more days after the onset (see Chapter 32B) may reveal low-amplitude maximum CMAPs. No evidence of significant demyelination-related motor conduction slowing or block exists. Sensory nerve action potentials (SNAPs) are normal. EMG examination in the acute phase shows profuse axon loss in the form of positive sharp waves and fibrillation potentials. In addition, fasciculations may be prominent. As motor axon loss progresses, evidence of neurogenic motor unit potential changes may be detected. The CSF typically shows increased protein content with normal glucose and a pleocytosis, with polymorphonuclear cells predominating during the acute stages and lymphocytes predominating later in the disease. Identification of CSF poliovirus-specific immunoglobulin M (IgM) antibody allows a specific diagnosis. Stool or nasopharyngeal cultures are positive for poliovirus in nearly 90% of patients by the 10th day of illness. The diagnosis may also be established by documenting a fourfold or greater increase in serum antibody titer against poliovirus from the acute as compared to the convalescent phase. Polymerase chain reaction (PCR) is now the best technique to diagnose poliovirus subtype as well as determine if the illness is related to wild-type versus Sabin oral polio vaccine–induced disease.
Vaccination
Immunocompromised individuals and their unimmunized direct household contacts are at particular risk for this rare complication. Consequently, in Northern America, Japan, Europe, New Zealand, and Australia, where wild viral infection is almost completely eradicated, policy shifted to use of the Salk inactivated vaccine in immunization schedules (Alexander et al., 2004).
Postpolio Syndrome/Progressive Postpoliomyelitis Muscular Atrophy
In the United States alone, it is estimated that 250,000 to 640,000 people survived acute paralytic poliomyelitis; the last epidemic was in 1952. Many years after recovery from acute poliomyelitis, some patients experience progressive functional impairment, with muscle fatigue, pain, sleep disturbances, cold intolerance, depression, dysphagia, and dysarthria, called the post-polio syndrome. If progressive muscle weakness and wasting occurs in this setting, the term progressive postpoliomyelitis muscular atrophy (PPMA) is used. The reported incidence of postpolio syndrome/PPMA among polio survivors ranges from 28.5% to 64%; no accurate estimate of the incidence exists. By definition, patients with postpolio syndrome/PPMA have recovered from acute poliomyelitis, and the disease course has been stable for at least 10 years after the recovery (Box 74.4).
Box 74.4 Characteristic Features of Postpolio Syndrome/Progressive Postpoliomyelitis Muscular Atrophy
EMG, Electromyography; PPMA, postpoliomyelitis muscular atrophy.
Treatment
No specific pharmacotherapy for postpolio syndrome exists. Care focuses on symptom relief (Gonzalez et al., 2010). A randomized controlled trial of intravenous immunoglobulin (IVIG; 2 courses of IVIG at a dose of 90 g per course over 3 days, with a 3-month interval) reported a significant improvement in muscle strength but not quality of life in 135 patients (Gonzalez et al., 2006). A need for further trials in larger patient groups exists.
West Nile Virus
West Nile virus (WNV) is an arthropod-borne flavivirus that cause epidemics of meningitis, encephalitis, and in some instances an acute polio-like flaccid paralysis. Approximately 80% of those who are infected are asymptomatic, and 20% develop a flulike illness (termed West Nile fever). Less than 1% present with neuroinvasive disease. Following the 1999 outbreak in New York, an epidemic spread across the North American continent and peaked in 2002/2003. Between 1999 and 2008, almost 29,000 confirmed and probable cases of WNV infection were received in the United States, and over 40% of cases were of the neuroinvasive type. The highest incidence of neuroinvasive WNV occurred in the western north-central United States and mountain states between the months of July and September (Lindsey et al., 2010). Epidemics also occurred in Israel, Italy, Russia, Romania, Hungary, Tunisia, the Sudan, the Caribbean, and Latin America. WNV is now endemic in North America and is the leading cause of arboviral encephalitis in the United States. WNV is a zoonotic pathogen that has crossed over from birds to humans, the latter serving as incidental hosts. The prime bridge vector is the Culex mosquito, and the spread of disease appears to have followed the migration patterns of bird populations (Kilpatrick et al., 2006). Human-to-human transmission can occur via blood transfusion, organ transplant, intrauterine exposure, and breastfeeding, hence the need to screen for the virus amongst blood donors. One of the most dramatic presentations is an acute asymmetrical flaccid paralysis in a febrile patient with or without meningitis, encephalitis, and cranial neuropathies (including hearing loss). Aching pains in affected limbs often accompany the paralysis, but actual sensory loss is not a feature. Respiratory failure and death may occur, and up to one-third of cases suffer bladder and bowel dysfunction. Recovery is very slow, and from the available evidence, incomplete. Other presentations include a GBS, multifocal chorioretinitis, pancreatitis, hepatitis, myocarditis, nephritis, and splenomegaly. Risk factors for death from WNV infection include chronic renal disease, an immunosuppressed state (e.g., in organ transplant recipients), the presence of encephalitis (versus meningitis) and old age (Murray, K., et al., 2006; Tyler et al., 2006). Detection of WNV-specific IgM antibody in the serum of a patient with abnormal CSF and acute neurological illness confirms the diagnosis. The diagnosis can also be confirmed by WNV-specific IgM antibody capture ELISA in CSF. PCR has also been developed to detect the virus (Tang et al., 2006). CSF protein is typically elevated (100 mg/dL or more and often higher in encephalitis versus meningitis), glucose is typically normal, and there are increased numbers of white cells (mean of about 220/mm3 with ≈ 45% neutrophils). The number of red cells is variable and may be higher in encephalitis (Tyler et al., 2006). Neuroimaging of the brain is usually normal, but that of the spinal cord may reveal increased signal in the anterior horns. Electrodiagnostic studies reveal an acute disorder of anterior horn cells, with acute loss of motor units in affected myotomes.
Multifocal Motor Neuropathy
A complete description of multifocal motor neuropathy is in Chapter 76. The condition is believed to be autoimmune in nature, and most cases have evidence of focal demyelination in the peripheral nerves (multifocal motor neuropathy with conduction block [MMNCB]) similar to that in chronic inflammatory demyelinating peripheral neuropathy. The clinical presentation, however, is with pure LMN involvement. The condition enters into the differential diagnosis of benign focal amyotrophy and the progressive muscular atrophy variant of ALS. It is important to search for this condition, since it is treatable by high-dose immunoglobulin infusions or other immunotherapy.
Benign Focal Amyotrophy
The terms benign focal amyotrophy, brachial monomelic amyotrophy, benign calf amyotrophy, Hirayama disease, or juvenile segmental muscular atrophy are used to describe disorders characterized by LMN disease clinically restricted to one limb. The etiology is unknown. Autopsy studies have shown the affected region of spinal cord flattened, the anterior horn markedly atrophied and gliotic, and a reduction in the numbers of both large and small motor neurons. Based upon neuroradiological studies, Hirayama, who established the disease entity, has proposed a mechanically induced limited form of ischemic cervical myelopathy, being the result of local compression of the dura and spinal cord against vertebrae during repeated neck flexion/extension, in turn due to disproportionate growth between the contents of the dural sac and the vertebral column (Hirayama, 2008; Hirayama and Tokamaru, 2000). However, surgical decompression has not altered the course of the disease, and this theory is no longer widely held. Another school of thought is that this is a segmental, perhaps genetically determined, SMA, but the actual cause is still unknown.
The disease usually begins in the late teens, but many cases can present in the fourth decade. More than 60% of patients are men. Although originally described in Indian and Japanese patients, the disorder is now recognizable around the world. The most common presentation is one of an idiopathic, slowly progressive, painless weakness and atrophy in one hand or forearm. The distribution of muscle weakness varies markedly from case to case, but a characteristic feature is that the condition remains limited to only a few myotomes in the affected limb. The most common pattern is unilateral atrophy of C7-T1 innervated muscles, with sparing of the brachioradialis (the “oblique atrophy” pattern). Muscle stretch reflexes are invariably hypoactive or absent in the muscles innervated by the involved cord segment but are normal elsewhere. UMN signs are not present, and if they are, one should consider the onset of ALS instead. Approximately 20% have hyperesthesia to pinprick and touch, usually located on the dorsum of the hand. The cranial nerves, pyramidal tracts, and the autonomic nervous system are normal. Weakness and atrophy may progress steadily for the initial 2 to 3 years, but most patients have stabilized within 5 years. The arm is the affected limb in approximately 75% of the patients and the leg in the remaining 25% (benign calf amyotrophy). Spread may occur to the contralateral limb in about 20% of cases (Gourie-Devi and Nalini, 2003), and rare patients later develop an ALS-like picture.
Spinal Muscular Atrophy
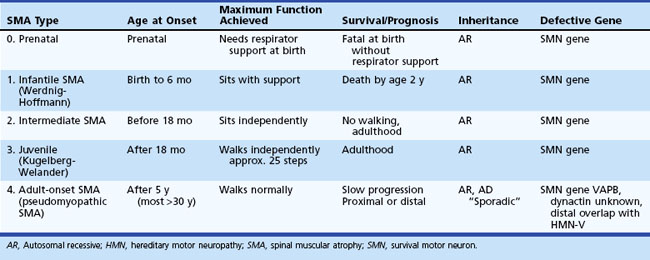
The SMA are a group of disorders caused by degeneration of anterior horn cells and, in some subtypes, of bulbar motor neurons. Almost all cases are genetically determined, with most being autosomal recessive due to homozygous deletions of the survival motor neuron (SMN) gene on chromosome 5. Traditionally, SMA is classified as one of the four types based on the age at onset: SMA type 1 (infantile SMA or Werdnig-Hoffmann syndrome), SMA type 2 (intermediate SMA), SMA type 3 (juvenile SMA or Kugelberg-Welander disease), and SMA type 4 (adult-onset SMA, pseudomyopathic SMA). A very severe prenatal form of SMA (type 0 SMA) can manifest prenatally with reduced fetal movements and respiratory distress at birth. It is also important to consider the maximum function that a child achieves in terms of sitting and walking; this is of prognostic significance. In the less severe forms of the disease, there can be periods where the child will improve or plateau, but long-term studies have demonstrated a net deterioration (Russman, 2007) (Table 74.2). The estimated incidence of infantile and juvenile recessive SMA is 1 in 6000 live births, with an approximate carrier frequency of 1 in 35 of the general population, making it a leading genetic cause of infant mortality. True adult-onset disease accounts for probably less than 10% of all cases of SMA, with an estimated prevalence of 0.32 in 100,000. The mean age at onset is the mid-30s but ranges from 20 to the late 40s. Up to 95% of all childhood cases are due to deletion of the survival motor neuron (SMN1, telomeric SMN, SMNT) gene located on chromosome 5q11.2-13.3. The remaining cases are due to small SMN mutations (rather than full deletions). SMN1 is located within an inverted gene duplication, the other half of which is occupied by the almost identical SMN2 (centromeric SMN, SMNC) gene. The SMN1 protein product is functionally absent in the vast majority (95%-98%) of cases of SMN-mutated SMA, and small amounts are present in the remaining few percent. The SMN2 protein is present in all patients, but the copy number can vary considerably. Only 1% to 2% of childhood-onset SMA is unrelated to the SMN locus on chromosome 5.
< div class='tao-gold-member'>
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


