and Robert E. Schmidt2
(1)
Sunnybrook and St Michael’s Hospitals, University of Toronto, Toronto, ON, Canada
(2)
Division of Neuropathology Department of Pathology, Washington University School of Medicine, St. Louis, MO, USA
14.1 Neuropathy Associated with Paraproteinemia
A significant association exists between the presence of circulating monoclonal immunoglobulins and peripheral neuropathy (NAP, neuropathy associated with paraproteinemia, is used throughout this chapter). In large series a paraprotein has been found in 5–10 % of otherwise idiopathic neuropathies; conversely, a neuropathy is seen in up to 58 % of patients with paraproteinemia (Read et al. 1978; Kelly et al. 1981a; Osby et al. 1982; Isobe and Osserman 1971; Vrethem et al. 1993; Walsh 1971; Smith et al. 1983; Ramchandren and Lewis 2012). In some cases the clinicopathological features of NAP suggests a causal relationship, while in others the significance of the association remains to be determined. The circulating paraprotein can be seen as part of several well-defined diseases, or may be present in isolation, with the frequency of neuropathy depending on the underlying disease, but, more importantly, on the particular type of paraprotein heavy or light chain. Table 14.1 lists the dysproteinemias associated with neuropathy (Kyle 1992).
Table 14.1
Diseases associated with paraproteinemia and neuropathy
Monoclonal gammopathies of unknown significance (MGUS) |
Multiple myeloma |
Waldenström macroglobulinemia |
Osteosclerotic myeloma and the POEMS syndrome |
Primary amyloidosis (can be seen in any of the above) |
Other lymphoproliferative diseases: |
Lymphoma |
Leukemia (CLL, hairy cell, myelomonocytic, others) |
Non-amyloid immunoglobulin deposition diseases |
Heavy chain disease |
Cryoglobulinemia (discussed separately) |
CANOMAD |
Paraproteins may consist of the entire immunoglobulin molecule or only the heavy or light chain. While there are some notable exceptions, clinical features of the paraproteinemic neuropathies are determined by the type of paraprotein (IgM vs. IgG or IgA, light chain vs. whole immunoglobulin) rather than with the associated disease. IgG is the most common class of protein in patients with benign paraproteinemia; however, IgM is more common in those patients with neuropathy (60 %, Luegetti et al. 2012), followed by IgG (30 %) and IgA (10 %). A small number of IgD paraprotein-associated neuropathies have been reported (Hansen et al. 1989; Merelli et al. 1986), but the infrequency of this finding and lack of adequate pathological study does not allow meaningful discussion of the “typical” syndrome or pathology. Thus, the individual diseases will be discussed briefly below, but clinical and pathological features will be discussed without reference to the underlying disease. Primary amyloidosis is discussed in Chap. 15.
14.1.1 Clinical Manifestations
14.1.1.1 Monoclonal Gammopathy of Unknown Significance (MGUS)
MGUS refers to a clonal expansion of B-cell lineage cells (lymphocytes and plasma cells) that secrete a monoclonal immunoglobulin (Caers et al. 2013). This is the most frequent dysproteinemia associated with peripheral neuropathy (recently reviewed in Ramchandren and Lewis 2012; Nobile-Orazio 2010; Kyle and Rajkumar 2005). “Benign” paraproteinemia is found in 1–3 % of unselected patients over the age of 50 and increases in an age-dependent fashion (Kohn 1976). The diagnostic criteria of MGUS comprise a level of monoclonal protein below 30 g/L, normal renal function, normal calcemia and hemoglobin level, bone marrow plasmacytosis below 10 %, and absence of bone osteolytic lesions, or systemic disease. The most common type of M-protein is IgG followed by IgM and then IgA. Monoclonal IgM can later transform into lymphoproliferative disorders such as Waldenström macroglobulinemia (WM), B-cell non-Hodgkin lymphoma, and chronic lymphocytic leukemia, while IgA and IgG paraproteinemias transform into MM (17 % in 10 years and 33 % by 20 years) (Luegetti et al. 2012). Light chain MGUS is a unique subtype of MGUS in which the secreted protein lacks the immunoglobulin heavy chain component (Caers et al. 2013; Lubimova et al. 2012).
Epidemiological evidence suggests an association between MGUS and neuropathy. Peripheral neuropathy is seen in 5–50 % of patients with MGUS (Kahn et al. 1980; Osby et al. 1982; Isobe and Osserman 1971; Nobile-Orazio et al. 1992). Conversely MGUS is found in 3.2–5.7 % of patients with otherwise cryptogenic peripheral neuropathy, severalfold the age-matched population prevalence (Sherman et al. 1984; Kelly et al. 1981a). Neuropathy is frequently the first manifestation of MGUS (Johansen and Leegaard 1985). Patients with MGUS and neuropathy demonstrate an IgM paraprotein far more frequently than patients with MGUS without neuropathy (Gosselin et al. 1991; Kahn et al. 1980; Yeung et al. 1991; Vrethem et al. 1993; Johansen and Leegaard 1985; Nobile-Orazio et al. 1992). About 50–90 % of IgM paraproteins associated with neuropathy have specificity for myelin-associated glycoprotein (MAG) and/or sulfate-3-glucuronyl paragloboside (Gosselin et al. 1991; Nobile-Orazio et al. 1992; Kelly 1990; Vital et al. 1989; Yeung et al. 1991). The majority of patients with anti-MAG antibody have no associated malignancy. The association between IgA and IgG paraproteins and neuropathy is not as impressive and often may represent the chance co-occurrence of two not-uncommon situations in the elderly (Smith et al. 1983; Kelly et al. 1981a; Nobile-Orazio et al. 1992; Read et al. 1978).
14.1.1.2 Multiple Myeloma
Multiple myeloma (MM) is distinguished from MGUS by the presence of a malignant proliferative clone of plasma cells, as demonstrated by bone marrow biopsy, high or rising paraprotein levels, or multisystem disease (renal, bone marrow). Symptomatic neuropathy (IgG kappa the most commonly associated paraprotein) has been described in 3.5–10 % of patients (Silverstein and Doniger 1963; Walsh 1971). As many as 40 % of such cases may be accounted for by primary amyloidosis (Kelly et al. 1981b), and others are caused by malignant myeloma nerve infiltration (Barron et al. 1960). However, in the majority of cases the cause of the neuropathy is not identified. Subclinical neuropathy is present in up to 40 % of patients (Silverstein and Doniger 1963; Walsh 1971; Kelly et al. 1981a). When it occurs, neuropathy is often an initial manifestation of the disease (Kelly et al. 1981a; Ramchandren and Lewis 2012). IgM paraprotein rarely occurs with MM.
14.1.1.3 POEMS Syndrome and the Osteosclerotic Myeloma
Osteosclerotic myeloma (OSM) is distinguished from multiple myeloma by the absence of anemia or marrow plasmacytosis and the presence of osteosclerotic bone lesions. A strong but not invariable association is seen with the POEMS syndrome of polyneuropathy, organomegaly (liver and spleen), endocrinopathy (thyroid, gonadal), M-protein, and skin changes (hyperpigmentation, hypertrichosis, edema). The diagnosis is confirmed by demonstrating clonal proliferation of plasma cells at the site of a sclerotic bone lesion (Kelly et al. 1983). Circulating paraproteins (typically IgG or IgA lambda), found in 93 % of patients with OSM, almost invariably have a λ light chain (Kelly et al. 1983). Osteosclerotic myeloma is unique in its strong association with neuropathy, which is found in at least 50 % of patients and is almost always the presenting manifestation, with subsequent investigation leading to the correct diagnosis (Kelly et al. 1983). OSM is also unique in that removal or irradiation of a localized lesion may result in clinical improvement (Kelly et al. 1983). Most reported cases of solitary plasmacytoma with neuropathy have shown osteosclerotic bone lesions (Read and Warlow 1978).
Depending on how the syndrome is defined, 5–45 % of patients with POEMS have no bone lesions, and 14 % of those who do have bone lesions have no sclerotic changes (Nakanishi et al. 1984; Miralles et al. 1992). Similarly, a paraprotein is not found in 13–25 % of patients with POEMS, although it may appear after a follow-up period (Nakanishi et al. 1984; Miralles et al. 1992). An association also exists between OSM, POEMS, and angiofollicular lymph node hyperplasia (Castleman disease), an uncommon nonmalignant disorder that occurs as either a localized or a multicentric form, which may include other systemic signs with features of POEMS. The complexity of associated lesions has resulted in redefinition and updating the syndrome (Dispenzieri 2011).
14.1.1.4 Waldenström Macroglobulinemia
WM is a B-cell malignancy characterized by a lymphoplasmacytic infiltration into the bone marrow and a monoclonal immunoglobulin M (IgM) protein in serum (Hodge and Ansell 2013). It differs from IgM MGUS on the amount of circulating paraprotein (>30 g/L in WM) and by the finding of either a 6q21 deletion or a mutation in the MYD88 gene. While some patients with WM have no symptoms at diagnosis, others present with anemia, retinopathy, bleeding, or neurological complaints. Neuropathy has been reported in 10–50 % of these patients and may precede or follow the systemic manifestations (Nobile-Orazio et al. 1987; Meier et al. 1984; Ramchandren and Lewis 2012). The circulating paraprotein (most commonly IgM kappa) in WM patients is associated with or without anti-MAG antibodies. Electrophysiological studies are indicative of demyelination.
14.1.1.5 Immunoglobulin Deposition Disease
Light chain deposition disease (LCDD) and light and heavy chain deposition disease (LHCDD) are rare conditions in which monoclonal light and/or heavy chains deposit diffusely in peripheral nerves. The light microscopic features are identical to amyloid but lack congophilia (Randall et al. 1976; Buxbaum 1992; Figueroa et al. 2012; Luigetti 2010). This amyloid-like deposition polyneuropathy occurs in patients having an IgM, kappa or lambda, monoclonal gammopathy, established WM, or MM. IgG or isolated light chains are most commonly seen in the systemic form of the disease.
14.1.1.6 CANOMAD
The constellation of chronic ataxic neuropathy with ophthalmoplegia, M-protein, cold agglutinins, and anti-disialosyl antibodies constitutes a rare disorder designated CANOMAD. The IgM proteins form antibodies against disialylated gangliosides.
14.1.1.7 Other Paraproteinemias and Lymphoproliferative Diseases
A few cases of solitary plasmacytoma have been associated with neuropathy, but most are best considered with osteosclerotic myeloma (Read and Warlow 1978). A circulating paraprotein, most often IgM, is seen in 3–18 % of patients with lymphoma, more frequently in cases with diffuse rather than nodular histology (Kyle and Garton 1987).
14.1.1.8 Neuropathic Manifestations
The neuropathy associated with paraproteinemia (NAP) is usually distal sensorimotor, symmetrical, of variable severity, and progresses over months to years. However, the syndrome can resemble polyradiculopathy or even mononeuritis multiplex (this may be a prominent picture in some patients with WM), may have predominantly motor or predominantly sensory presentations, and can progress in a subacute, stepwise, or relapsing and remitting fashion (Mamoli et al. 1991; Kusunoki et al. 1989; Rowland et al. 1982; Lamarca et al. 1987; Case records MGH 1993; Yeung et al. 1991; Gosselin et al. 1991). Reflex loss is usually prominent, CSF protein is most often increased, and nerve conduction studies usually reveal demyelinating or mixed features (Gosselin et al. 1991; Yeung et al. 1991; Meier 1985; Kelly 1983). The neuropathy associated with osteosclerotic myeloma and the POEMS syndrome is somewhat distinct in its motor predominance, greater severity, and more consistent demyelinating features (Kelly 1983). The clinical picture of the chronic and progressive polyneuropathy of CANOMAD is marked by sensory ataxia and areflexia, with sparing of distal motor strength and function (Ramchandren and Lewis 2012).
From the study of the small number of immunoglobulin deposition disease cases published, one discerns two clinical syndromes of peripheral nerve involvement: a progressive (distal to proximal), painful, asymmetric sensorimotor axonal polyneuropathy and a mononeuropathy multiplex-like picture (Leschziner et al. 2009; Luigetti et al. 2010). The relation between typical chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) without a paraprotein (Chap. 9) and a CIDP-like picture associated with a circulating paraprotein, seen in 8–30 % of “CIDP” cases (Barohn et al. 1989; Bromberg et al. 1992), is problematic. Some authors exclude the latter group of patients from the definition of CIDP (Dalakas and Engel 1981; Dyck et al. 1975; Kyle 1992) or classify them separately (Ad Hoc subcommittee 1991). Clinically the two syndromes are far more similar (manifestations, electrophysiology, CSF parameters, and response to treatment) than they are different, regardless of paraprotein immunoglobulin class (Bleasel et al. 1993; Bromberg et al. 1992; Yeung et al. 1991; Kyle 1992; Gosselin et al. 1991), and the histological distinction between the two is also potentially difficult (vide infra).
14.1.1.9 Treatment
Treatment for patients with circulating paraprotein-associated neuropathy includes plasma exchange (Sherman et al. 1984; Dyck et al. 1991; Meier et al. 1984), intravenous immune globulin (Cook et al. 1990) steroids, and cytotoxic alkylating drugs (Dalakas and Engel 1981; Smith et al. 1987; Yeung et al. 1991). All have been reported to improve clinical status, but results are unpredictable. In patients with a localized plasmacytoma or osteosclerotic myeloma, there are reports of benefit with resection or local irradiation, bortezomib, and stem cell therapy, although “cure” is unlikely (Broussolle et al. 1991; Nakanishi et al. 1984; Miralles et al. 1992; Kelly 1983; Read and Warlow 1978; Ramchandren and Lewis 2012). Plasmapheresis is more effective with IgG/IgA paraproteinemic neuropathies. Monoclonal antibody (rituximab) against the CD20 surface antigen is the most promising drug for anti-MAG neuropathy (Dalakas 2010).
14.1.2 Pathology
14.1.2.1 General Considerations
The pathological alterations observed in most cases of IgG or IgA paraprotein and some cases with an IgM paraprotein are generally nonspecific, except for increased periodicity of myelin lamellae; widely spaced myelin is highly suggestive of a subset of IgM paraproteins with activity against MAG and other myelin antigens, while uncompacted myelin is strongly associated with the POEMS syndrome. Full investigation of a patient with neuropathy may reveal the presence of a paraprotein, cryoglobulins, or a sclerotic bone lesion. In such cases, biopsy results will not modify management or give useful prognostic information unless a different diagnosis is found, perhaps vasculitis or amyloidosis. Thus we do not recommend nerve biopsy for patients with a known paraprotein unless the picture is atypical and suggestive of a different diagnosis. A simultaneous muscle biopsy will increase the diagnostic yield in both vasculitis and amyloidosis.
The pathological findings in NAP depend more on the heavy chain type (IgG and IgA vs. IgM) than on the underlying disease (MGUS, MM, OSM, or WM). The discussion below is organized accordingly. Cryoglobulinemia is discussed separately later in this chapter.
14.1.2.2 Light Microscopy
In NAP, nerves not uncommonly show focal, epineurial, and endoneurial, infiltrates of lymphocytes and macrophages and, less frequently, plasmacytoid cells are observed (arrows, Fig. 14.1a, b) (Vital et al. 1982; Julien et al. 1984a; Luigetti 2012), although multiple myeloma can cause massive nerve infiltration (Barron et al. 1960). Some reports have documented perineurial widening due to thickening and vacuolation of perineurial cells (Iwashita et al. 1974) or excessive spacing between perineurial layers (Smith et al. 1983). A circulating paraprotein is rarely associated with necrotizing vasculitis (Gherardi et al. 1989; Panegyres et al. 1990).
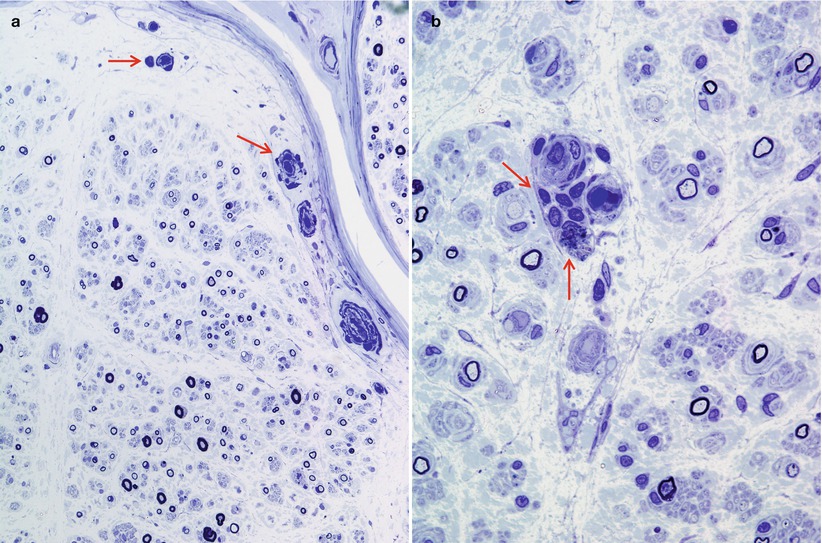
Fig. 14.1
Lambda chain MGUS NAP: semithin sections reveal rare endoneurial perivascular mononuclear cells (arrows, a, b), moderate dropout of fibers, and numerous thinly myelinated elements (a, b 1 μ thick plastic sections, 1,000×)
The spectrum of axonal and demyelinating changes seen in IgG, IgA, and IgM paraprotein-associated neuropathies overlaps considerably. Demyelinating changes such as active myelin breakdown, denuded axons, and thinly myelinated fibers are, on average, more prominent in IgM paraprotein-associated neuropathy, especially if the IgM paraprotein has activity against MAG (Meier 1985; Smith et al. 1987; Yeung et al. 1991; Walsh 1971; Vital et al. 1989; Simmons et al. 1993; Powell et al. 1984). Active axonal degeneration and debris-filled macrophages can be present, but more common is an overall reduction in the number of fibers. Examination of a patient with IgM NAP early in her disease (Fig. 14.2a, c and 16 years later (Fig. 14.2b, d) shows the progression of axonal loss, presented as axon number vs. size distribution (Fig. 14.3) and development of onion bulbs. Onion-bulb formations, as well as redundant myelin loops and/or abnormally thick myelin, are most commonly observed in IgM paraprotein-associated neuropathies (Vital et al. 1985a, b, 1989; Nardelli et al. 1981; Meier et al. 1983; Smith et al. 1983; Rebai et al. 1989; Jacobs and Scadding 1990; Lach et al. 1993). Regenerating clusters may be seen. Unmyelinated fibers may be depleted, but this is usually overshadowed by the loss of myelinated axons (Walsh 1971; Vital et al. 1989). Strong evidence exists that segmental demyelination, at least in cases not associated with an IgM paraprotein, is secondary to axonal atrophy (Ohi et al. 1985).
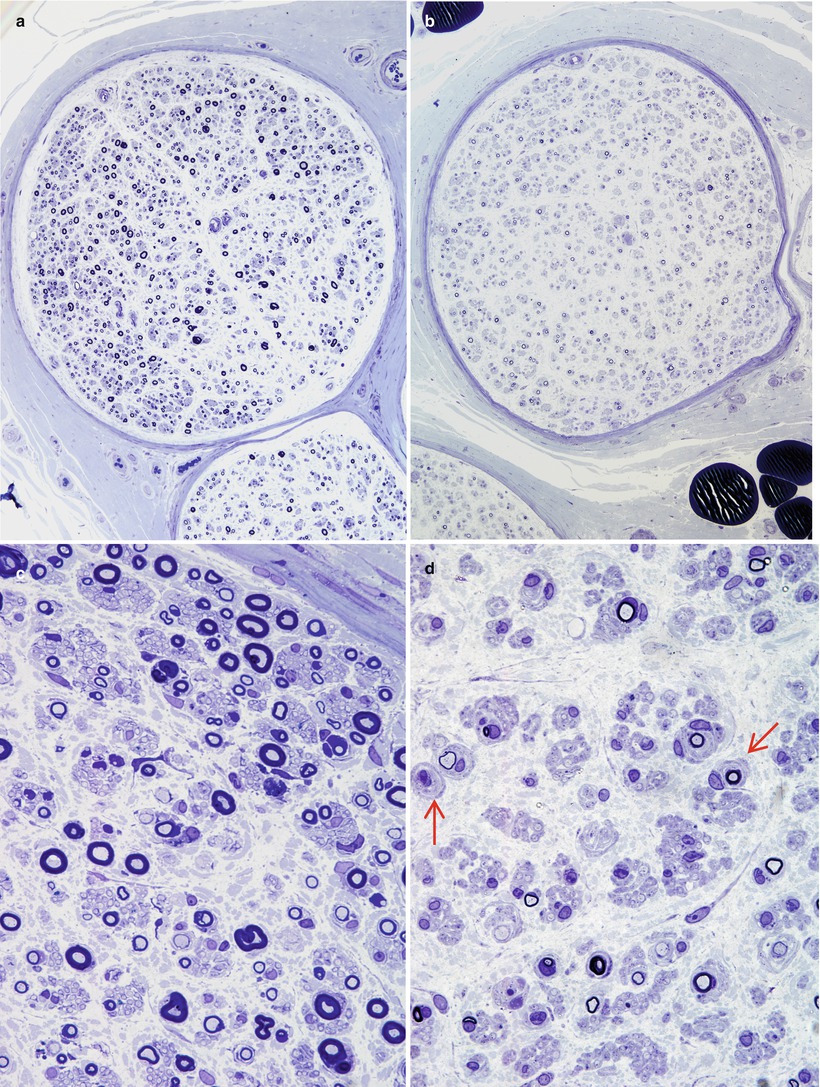
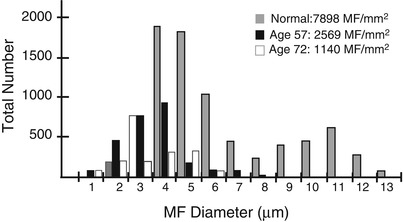
Fig. 14.2
IgM NAP: monoclonal gammopathy of 16 years’ duration. Sural nerve biopsies in 1978 (a, c) and in 1993 (b, d). Note progression in severity of axonal loss and myelin alterations, with onion-bulb formation (arrows d) (1 μ thick plastic sections, a, c 100×; b, d 600×)
Fig. 14.3
IgM NAP: fiber diameter-frequency histograms chronicle the slow but relentless loss of axons in this acquired neuropathy of 16 years’ duration
Nerve biopsies in the POEMS syndrome have revealed axonal degeneration, segmental demyelination, or both, with little or no inflammatory infiltration (Vital et al. 1994; Nakanishi et al. 1984; Kelly et al. 1983). Axonal degeneration predominated in the one case we have examined. Very few neuropathological studies of CANOMAD have been undertaken. In an autopsied case (McKelvie et al. 2013), there was severe dorsal column degeneration, dropout of ganglion cells in spinal ganglia, and endoneurial infiltration of clonal B-lymphocytes in cranial and peripheral nerves, dorsal roots, and cauda equina.
In immunoglobulin deposition disease, endoneurial, perivascular, and subperineurial deposition of PAS-positive (diastase-resistant) Congo red negative amorphous material has been reported (Dubas et al. 1987; Iwashita et al. 1974; Lamarca et al. 1987). The proteinaceous aggregates appear as hyaline eosinophilic pools associated with blood vessels and are bluish in toluidine blue-stained semithin plastic sections (Fig. 14.4a–c). While many cases exhibit immunoreactivity for kappa light chain, labeling for lambda is also on record (Leschziner et al. 2009). Using mass spectrometry of microdissected tissue, a recent paper document in three patients that the amyloid-like endoneurial amorphous aggregates result from the deposition of the whole IgM molecule (Figueroa et al. 2012). This contrasts with primary amyloidosis, where the intraneural deposits consist entirely of Ig light chains.
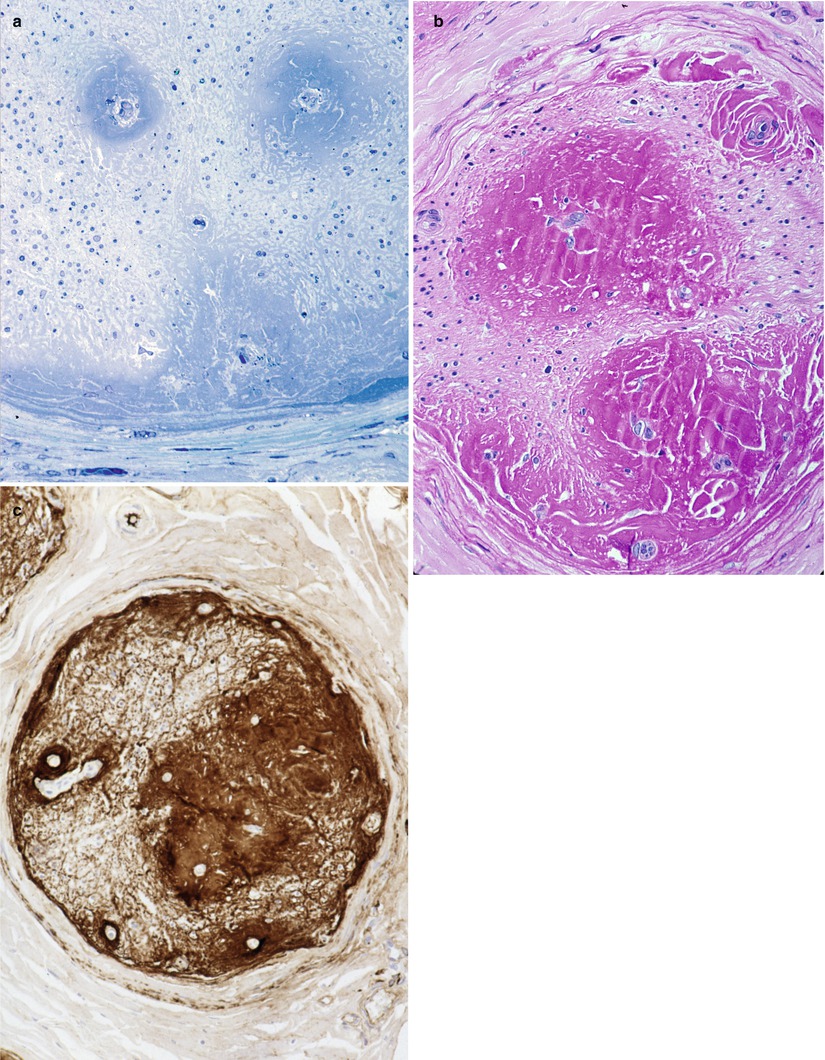
Fig. 14.4
Kappa chain NAP: (a) This fascicle shows a desolate endoneurium with no residual nerve fibers. Note subperineurial and perivascular deposition of amorphous material which is PAS positive (b) and Congo red negative. Intense immunoreactivity for kappa light chain is obtained (c) (a: 1 μ thick plastic sections 200×; b, c: paraffin 100×)
14.1.2.3 Electron Microscopy
An increase in myelin periodicity has been the most discussed histological feature in paraproteinemic neuropathies. The distinction between uncompacted myelin (UCM) and widely spaced myelin (WSM) is characterized by alterations in the patterns of periodicity (vide infra). Additional details of the ultrastructural features and possible mechanism of formation are provided in Chap. 5. Trauma during nerve handling, especially traction, can cause splitting of the delicate myelin sheath, which may mimic alteration in myelin periodicity. This usually manifests as a wavy, irregular separation of myelin lamellae which should not be difficult to distinguish from abnormalities of myelin periodicity (Fig. 7.1).
WSM has been described more frequently and is due to lack of apposition of the outer (extracellular) aspects of the Schwann cell membrane. The intraperiod line can no longer be identified, as the normal 2–4 nm separation between paired membranes is increased to 20–30 nm (Fig. 14.5a–d) and represents a virtual extracellular space. Intrusion of anti-MAG antibodies opens the intraperiod line, producing a wide space between the myelin lamellae (20–30 nm) resulting in myelin breakdown and remodeling (Kawagashira et al. 2010). Although there is a tendency for WSM to be seen in the outer layers of the myelin sheath, it has been described in the inner layers or throughout the entire thickness of the sheath. WSM is seen in 50–90 % of an IgM paraprotein-associated neuropathies, and anti-MAG antibody activity is usually, but not invariably, found in these cases (King and Thomas 1984; Vital et al. 1989; Yeung et al. 1991). Unusual configurations of redundant myelin (Fig. 14.6) may be found involving WSM. Two cases with WSM have been reported in the setting of IgG or IgA paraprotein-associated neuropathy (Powell et al. 1984; Vital et al. 1989).
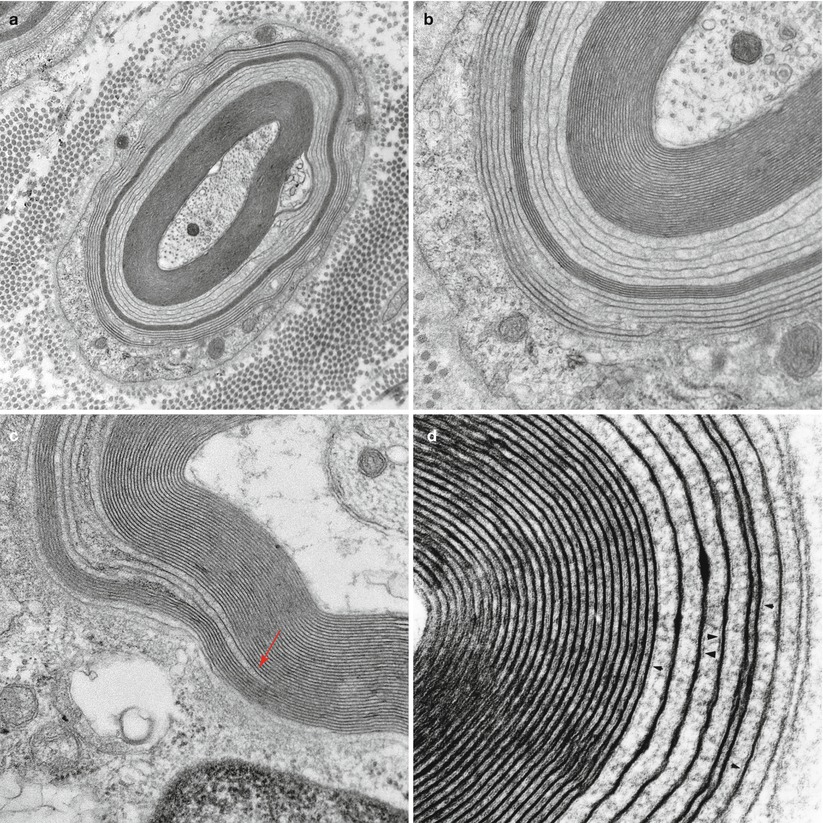
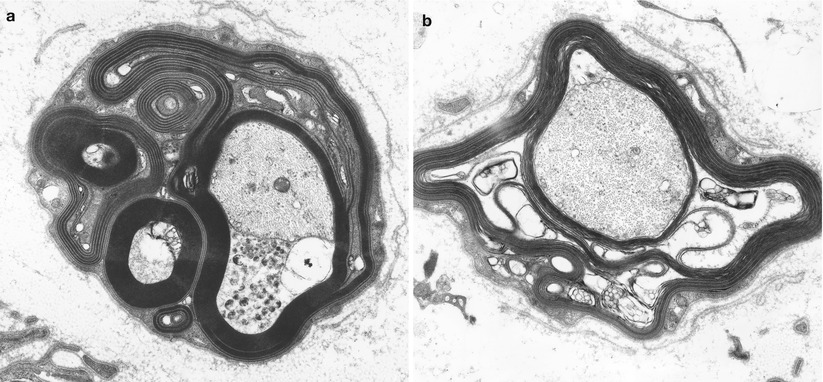
Fig. 14.5
IgM NAP: (a–d) Widely spaced myelin (WSM). (a, b) This myelinated axon shows the classic features of widely spaced myelin involving some of the lamellae, seen at higher magnification in (b). (c) Point of myelin separation (arrow, c). (d) Residual fragments of the intraperiod line are seen in the areas of WSM (arrowheads, d) (a, 20,000×; b, 50,000×; c, 60,000×; d, 137,000×)
Fig. 14.6
IgM NAP: Unusual configurations of redundant myelin with WSM are shown (a). Note intramyelinic splitting and vesicular degeneration (b) (a, 14,910×; b, 22,800×)
A different pattern of altered myelin periodicity has been designed uncompacted myelin (UCM). In UCM the major dense line is not formed because the inner (cytoplasmic) aspects of the Schwann cell membrane do not fuse (Fig. 14.7a–c). This results in an axon wrapped in spirals of Schwann cell cytoplasm. Vital et al. (1994) found this alteration in 1–16 % of internode cross sections in 19 of 22 biopsies in patients with POEMS syndrome, while Ohnishi (1984) indicated its presence in 3–8 % of cross sections in “over half” of nine cases of POEMS syndrome. A circulating paraprotein is usually, but not invariably, present, usually IgG or IgA (Bergouignan et al. 1987; Gherardi et al. 1988; Ohnishi and Hirano 1981; Vital et al. 1985b, 1994). Macrophage-mediated myelin stripping, generally regarded as the hallmark of the inflammatory demyelinating neuropathies GBS and CIDP, is occasionally seen in IgM (Vital et al. 1991b) and IgG (Bleasel et al. 1993; Pollard et al. 1983) paraprotein-associated neuropathies.
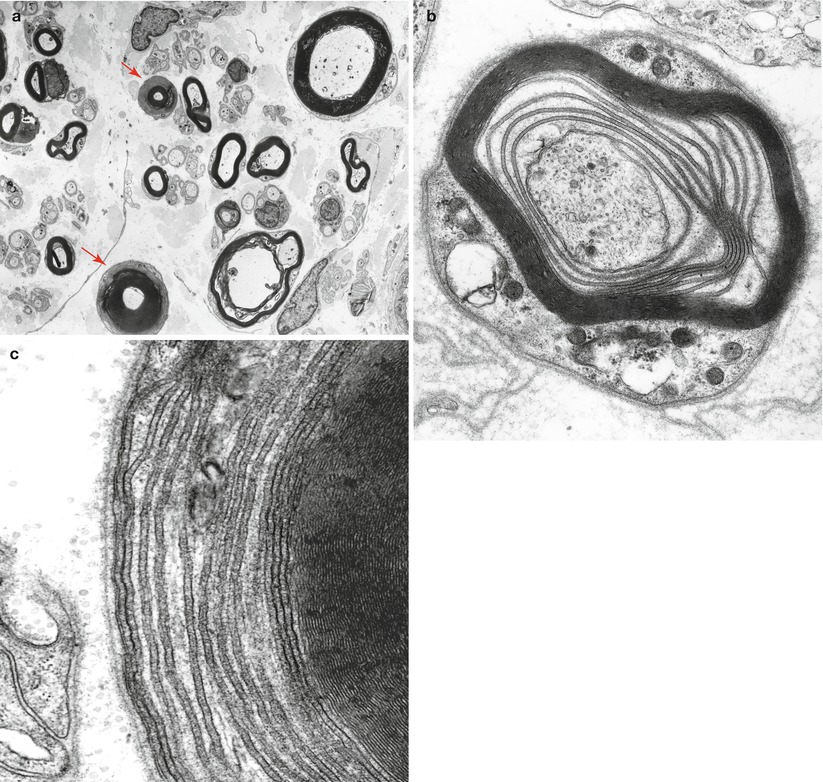
Fig. 14.7
Uncompacted myelin (UCM) two axons show uncompacted myelin (arrows, a). At higher magnification loss of myelin compaction is seen which shows separation at the major dense line
Cases of IgM paraprotein-associated neuropathy having the features of immunoglobulin deposition disease show focal or diffuse deposition of granular material, lacking the distinctive “rigid” fibrillar ultrastructure of amyloid, in the endoneurium or around endoneurial vessels (Fig. 14.8a, b) (Dubas et al. 1987; Iwashita et al. 1974; Lamarca et al. 1987; Meier et al. 1984; Vital and Vital 1993). This material does not stain histochemically for amyloid, but will immunostain for light and/or heavy chain of the circulating paraprotein. At times this deposited material seems to cause Schwann cell and vascular injury (Lamarca et al. 1987; Meier et al. 1984; Vital and Vital 1993). Endoneurial accumulation of amorphous or granular electron opaque material and small bundles of 10–18 nm diameter filaments has been also reported in IgM paraproteinemia with anti-chondroitin sulfate specificity (Yee et al. 1989; Sherman et al. 1983). Subperineurial and endoneurial elaunin collections have at times been mistaken for endoneurial immunoglobulin deposition (Fig. 14.9a) (Chazot et al. 1976; Carrier et al. 1978; Fitting et al. 1979). A patient with axonal neuropathy and an IgG kappa paraprotein whose peripheral nerve showed “immunotactoid-like” material intra-axonally and in endoneurium (Moorhouse et al. 1992) has been reported. This material was amorphous on light microscopy but stained positively for IgG. Ultrastructurally, the deposits were composed of straight or slightly curved single, bi-, or tri-layered tubular structures 50–300 nm in diameter. The neural microvasculature appeared damaged.
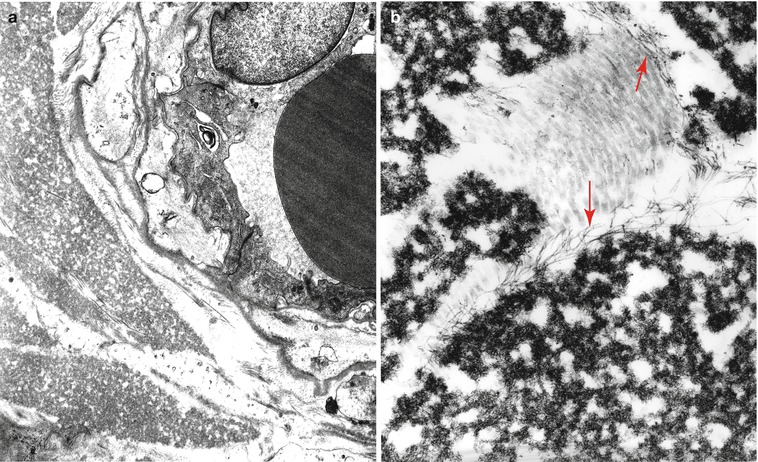
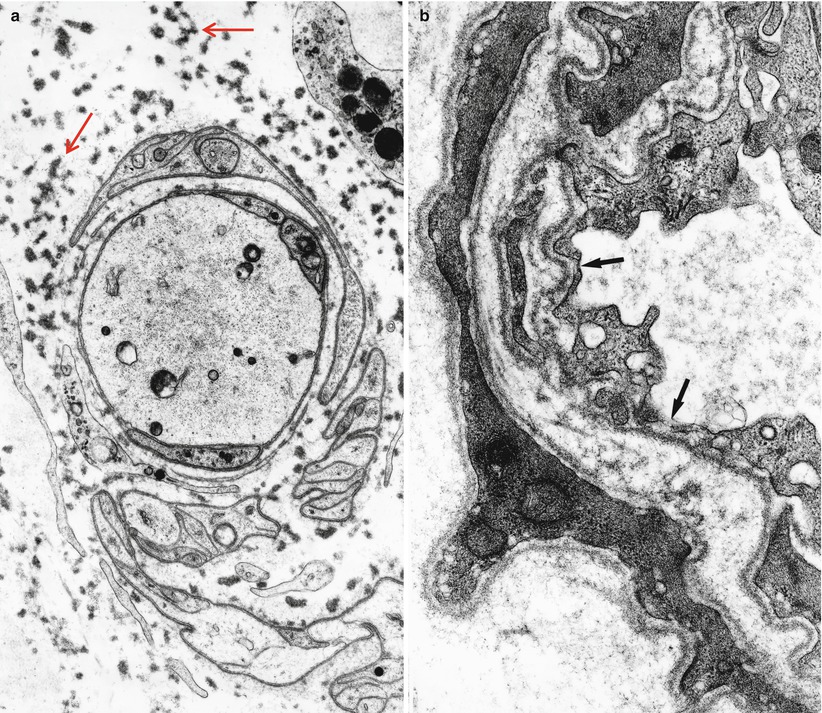
Fig. 14.8
Kappa chain NAP: (a) Ultrastructure demonstrates a perivascular granular deposit without features of amyloid. (b) Endoneurium shows collagen fibers, oxytalan filaments (arrows), and granular osmiophilic material considered typical of non-amyloid light chain (same case as Fig. 14.4). (a: 15,000×; b 33,800×)
Fig. 14.9
IgM NAP: Endoneurial deposition of elaunin is prominent (arrows, a). “Porous” endothelium is noted in endoneurial vessels (arrows, b) (a, 8,800×; b, 32,200×)
Microangiopathy has been emphasized by Powell et al. (1984) in neuropathy associated with IgG and IgM paraproteinemia. These authors demonstrated swollen capillary endothelial cells filled with microfilaments. A similar finding was seen in 1 of 31 biopsies in IgM gammopathy and neuropathy (Vital et al. 1989). However, a degree of endothelial cell hypertrophy, reduplication of basement membrane, and filamentous accumulations in endothelial cells are all common nonspecific findings of chronic neuropathy. Definitive evidence of an impaired blood nerve barrier, such as fenestration, gaps between endothelial cells, or excessive pinocytotic vesicle activity, has also been observed (Fig. 14.9b) (Lach et al. 1993; Powell et al. 1984; Meier et al. 1984).
Ultrastructural examination allows close assessment of axonal viability and provides evidence of primary demyelination when degenerating myelin is seen around healthy axons. Some authors (Jacobs and Scadding 1990; Ohi et al. 1985; Mendell et al. 1985) have emphasized axonal atrophy as evidenced by the finding of myelin inappropriately wide for axon diameter or retraction of the axons from the myelin sheath (Fig. 14.10). However, the former change is hard to separate from hypermyelination (commonly seen in IgM paraproteinemic neuropathy), while the latter may be artifactual. Such an atrophic appearance is currently thought to reflect intramyelinic edema. Quantitative studies by Ohi et al. (1985) support the hypothesis that axonal atrophy is present, as do autopsy findings in another case (Mendell et al. 1985). Evidence of unmyelinated fiber loss, with stacks of Schwann cell processes (Bands of Büngner) and collagen pockets, can often be seen, indicating that unmyelinated axons are not always spared (Vital et al. 1989).
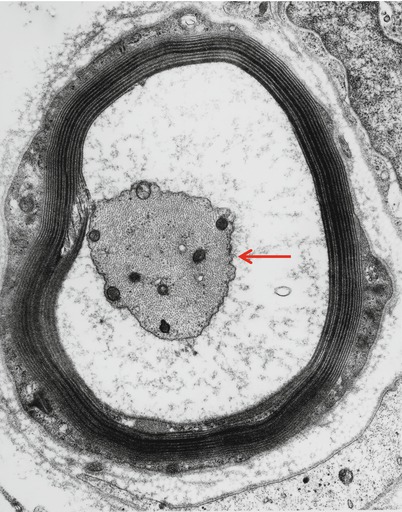
Fig. 14.10
IgM NAP: An adaxonal space resulting from intramyelinic edema separates a shrunken atrophic axon (arrow) from its myelin (14,560×)
14.1.2.4 Immunohistochemistry
Immunohistochemistry for the diagnosis of paraproteinemic neuropathy may give results that are inconsistent and difficult to interpret. Direct immunofluorescence of frozen tissue and immunoperoxidase staining of frozen or fixed tissue have been used in NAP to search for paraprotein binding to nerve (Fig. 14.11). Similarly, immunogold staining has conclusively demonstrated that IgM is found in the regions of widened myelin (Fig. 14.12) (Lach et al. 1993). Many reports demonstrate localization of the clonal immunoglobulin (heavy or light chain) to the myelin sheath (Mendell et al. 1985; Propp et al. 1975; Stefansson et al. 1983; Vital et al. 1982, 1989; Meier et al. 1983; Dellagi et al. 1983; Takatsu et al. 1985; Smith et al. 1983; Schenone et al. 1988; Dubas et al. 1987; Johansen and Leegaard 1985; Pollard et al. 1985; Yeung et al. 1991). When neuropathy is associated with an IgM paraprotein, this technique is positive in 40–80 % of cases (Dellagi et al. 1983; Dubas et al. 1987; Yeung et al. 1991), usually in the setting of a paraprotein with anti-MAG activity (Nobile-Orazio et al. 1987; Vital et al. 1989; Yeung et al. 1991; Takatsu et al. 1985). Myelin binding can be to the periphery of the sheath (Mendell et al. 1985; Pollard et al. 1985) or to its entire thickness (Smith et al. 1983). In IgG and IgA NAP, this finding is uncommon (Yeung et al. 1991; Dalakas and Engel 1981; Bailey et al. 1986; Bleasel et al. 1993; Sewell et al. 1981). Similar binding may be present in asymptomatic patients with a paraprotein (Dellagi et al. 1983) and in CIDP (Dalakas and Engel 1980). Indirect immunofluorescence using patient serum and animal or human nerve is often positive and can be performed on fixed or frozen material (Dellagi et al. 1983; Takatsu et al. 1985).
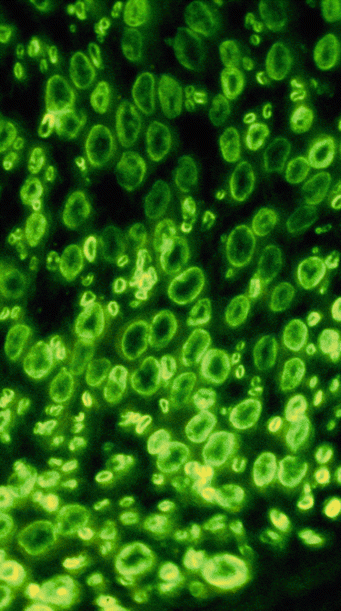
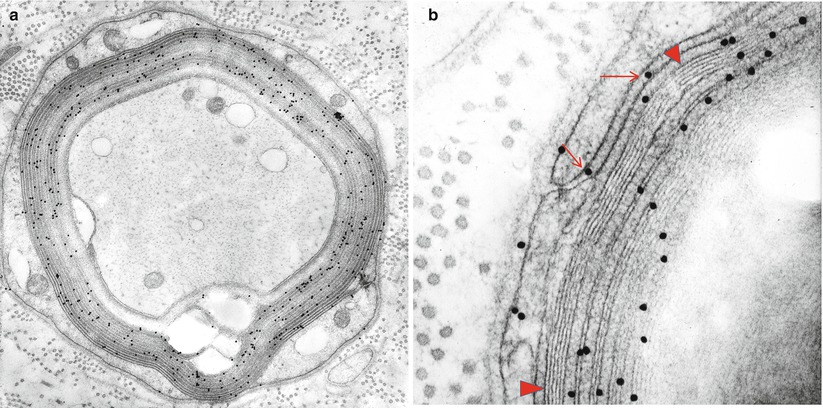
Fig. 14.11
Fluorescent anti-IgM antibody demonstrates immunolocalization of bound IgM to myelin sheaths of large and small myelinated axons (1,000×)
Fig. 14.12
IgM NAP: (a, b) Bound IgM is immunolocalized to WSM areas (arrows, b) and spares compacted myelin (arrowheads, b)
Positive perineurial anti-IgM immunostaining is a common but nonspecific finding (Propp et al. 1975; Johansen and Leegaard 1985; Yeung et al. 1991; Nobile-Orazio et al. 1987). However, positive endoneurial immunostaining for IgM is an important observation (Vital et al. 1989; Takatsu et al. 1985; Jonsson et al. 1988; Chazot et al. 1976; Vital and Vital 1993). A few such cases have been associated with anti-chondroitin sulfate IgM paraprotein-associated neuropathy (Sherman et al. 1983; Yee et al. 1989), typically presenting with an axonal sensory neuropathy and showing diffuse staining of endoneurial connective tissues. Focal areas of positivity likely correspond to the local immunoglobulin deposits discussed above (Iwashita et al. 1974; Chazot et al. 1976; Vital and Vital 1993). Light chain deposits may also be detected (Fig. 14.13). In IgG and IgA paraprotein-associated neuropathies, immunohistochemistry is of little value because IgG and, to a lesser extent, IgA can pass through an intact blood nerve barrier, so their presence in endoneurium in the setting of increased serum levels would not be surprising.
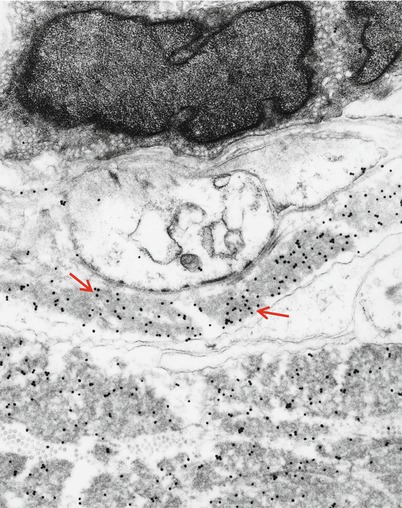
Fig. 14.13
Kappa chain NAP: Immunoelectronmicroscopy. Gold particles label kappa light chain composition of granular endoneurial deposits (same case as Fig. 14.4) (19,170×)
14.1.3 Pathogenesis
Although the clinical syndromes and pathology of the various paraprotein-associated neuropathies overlap, the IgM-associated neuropathies are somewhat distinct clinically and pathologically and will be considered separately. Only in IgM paraprotein with activity against MAG and related antigens has a causal relation between paraprotein and neuropathy been convincingly demonstrated.
14.1.3.1 IgM “Anti-MAG” Paraprotein-Associated Neuropathy
Reactivity toward MAG is seen in 50–90 % of IgM paraproteins associated with neuropathy (Nobile-Orazio et al. 1989; Quarles 1989; Kelly 1990; Vital et al. 1989; Yeung et al. 1991), and there is a strong correlation between the presence of anti-MAG IgM in myelin and the presence of a demyelinating neuropathy with widened myelin lamellae (Vital et al. 1989; Mendell et al. 1985; Monaco et al. 1990; Yeung et al. 1991). The use of immunogold technique has conclusively demonstrated the localization of IgM and light chain to the separated myelin lamellae in one case (Lach et al. 1993). The central question is whether presence of the antibody in myelin is an epiphenomenon or a primary event in the neuropathy.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


