and Robert E. Schmidt2
(1)
Sunnybrook and St Michael’s Hospitals, University of Toronto, Toronto, ON, Canada
(2)
Division of Neuropathology Department of Pathology, Washington University School of Medicine, St. Louis, MO, USA
Systemic malignancy is often associated with polyneuropathy. In this chapter we discuss neuropathy due to tumor infiltration of the peripheral nerve and neuropathy associated with the “paraneoplastic” remote effects of malignancy. We exclude peripheral nerve disease caused by clearly identified systemic metabolic disturbances such as renal failure or malabsorption or occurring as a complication of chemotherapy (see Chap. 18). The neuropathy associated with circulating paraproteins or cryoglobulins is discussed in Chap. 14.
16.1 Paraneoplastic Neuropathy
Paraneoplastic neuropathy has been the subject of recent, extensive reviews (Giometto et al. 2010; Grisold et al. 2013; Graus and Dalmau 2013; Koike and Sobue 2013). Formally defined, paraneoplastic neurological syndromes are rare neurological complications occurring in less than 1 % of people with malignancy which are not due to direct tumor infiltration or metastases, ischemia, metabolic deficits, nutritional deficits, or side effects of chemotherapy (Graus et al. 2004). Paraneoplastic conditions involving the central and peripheral nervous systems are thought to result from an autoimmune response to onconeural antigens shared by the cancer and the peripheral or central nervous systems. A large study (979 patients) performed by the Paraneoplastic Neurological Syndrome Euronetwork (PNSE) (Giometto et al. 2010) has developed criteria for the diagnosis of paraneoplastic neurological syndromes based on the earlier classification of Graus et al. (2004). A definite paraneoplastic syndrome is that arising in a patient with idiopathic neurological symptoms and seropositivity for any well-characterized autoantibodies (anti-Hu, Yo, CV2/CRMP-5, Ri, Ma2, or amphiphysin), regardless of whether an underlying malignancy is found. Other schemes require the cancer to develop within 5 years of the diagnosis of the neurological disorder. However, some cases may not, at the time of diagnosis, be definitively associated with an identifiable onconeural antibody (Rudnicki and Dalmau 2005; Molinuevo et al. 1999) and, as a result, are typically considered nonclassical or “possible” paraneoplastic syndrome. Using these criteria, classical paraneoplastic syndromes of the peripheral nervous system include two major entities: subacute sensory neuronopathy and chronic gastrointestinal pseudo-obstruction (Graus et al. 2004), both often associated with anti-Hu antibodies. In a small study of 20 patients with paraneoplastic neuropathy due to anti-Hu antibodies, neuropathy was classified as sensory in 70 % of patients, sensorimotor in 25 %, and motor in 5 % (Camdessanché et al. 2002). A variety of malignancies have been associated with paraneoplastic peripheral neuropathies, the lung most commonly (vide infra).
Thus, peripheral neuropathies associated with neoplasia include acute, subacute, and chronic sensory and sensorimotor neuropathies, neuropathy with vasculitis, and autonomic neuropathies. Vasculitis is more frequent with hematologic malignancies than with solid malignancies (Fain et al. 2007) and may respond to immunosuppressive therapy (Oh et al. 1991; Antoine et al. 1999).
16.1.1 Paraneoplastic Subacute Sensory Neuropathy (SSN)
According to the PNSE database (Giometto et al. 2010), one-third of patients with paraneoplastic syndromes show primary involvement of the peripheral nervous system, and, of those, subacute sensory neuropathy is the most common form (Graus et al. 2004; Giometto et al. 2010) and may anticipate the detection of neoplasia (Dalmau et al. 1992; Graus et al. 2001). Most commonly (66–76 % of cases), small cell lung cancer (SCLC) is the associated malignancy, but squamous and anaplastic lung cancers, Hodgkin’s disease, breast or ovarian cancer, sarcoma, lymphoma, thymoma, and adenocarcinomas have also been described, and single case reports document other malignancies in this setting (Dalmau et al. 1992; Smith 1992; Horwich et al. 1977; Gozzard and Maddison 2010).
16.1.1.1 Clinical Manifestations
Subacute sensory neuropathy (SSN) is the most common paraneoplastic peripheral neuropathy (24 % of patients in a large European series, Giometto et al. 2010) and usually precedes diagnosis of the associated tumor, sometimes for as long as several years (Dyck et al. 1958; Dalmau et al. 1992; Chalk et al. 1992). Patients present with combinations of sensory ataxia, paresthesias, and limb pain, often with early involvement of the upper extremity, but with little or no weakness or wasting (although involvement of motor functions does not exclude SSN, Graus et al. 2004). Oki et al. (2007) divided paraneoplastic sensory neuropathy into patients with ataxia vs. those with pain which may reflect the size of targeted DRG neurons. Autonomic symptoms, such as urinary retention, orthostatic hypotension, or intestinal pseudo-obstruction, the last often a prominent early feature, may also occur in patients with subacute sensory neuronopathy (Oki et al. 2007). Autonomic features, present in 5 % of a large European series (Giometto et al. 2010) may herald a worse prognosis. A number of patients with autonomic involvement may have anti-Hu, anti-CV2/CRMP-5, or anti-Ach receptor antibodies (Lucchinetti et al. 1998; Yu et al. 2001; McKeon et al. 2009; Graus and Dalmau 2013). Progression occurs over days to weeks, often to a severity which precludes limb use. Concurrent paraneoplastic CNS involvement is common, including encephalopathy, cerebellar syndrome, or myelopathy. The CSF protein is typically elevated and the cell count usually normal. Electrical testing shows features of an axonal sensory neuropathy with normal or minimally altered motor conductions and EMG.
Survival from onset of neurological symptoms averages 2–3 years (Horwich et al. 1977), and death results as often from the neurological disease as from the underlying tumor (Dalmau et al. 1992). With a few exceptions, treatment appears to be ineffective, including that directed at the associated malignancy, but several patients with Hodgkin’s disease have experienced improvement with successful treatment of their malignancy (Smith 1992).
A clinically similar inflammatory sensory ganglionopathy can occur in the absence of systemic malignancy (Smith 1992). However, central nervous system features are absent, and the anti-Hu antibody (vide infra) is not detected.
16.1.1.2 Pathology
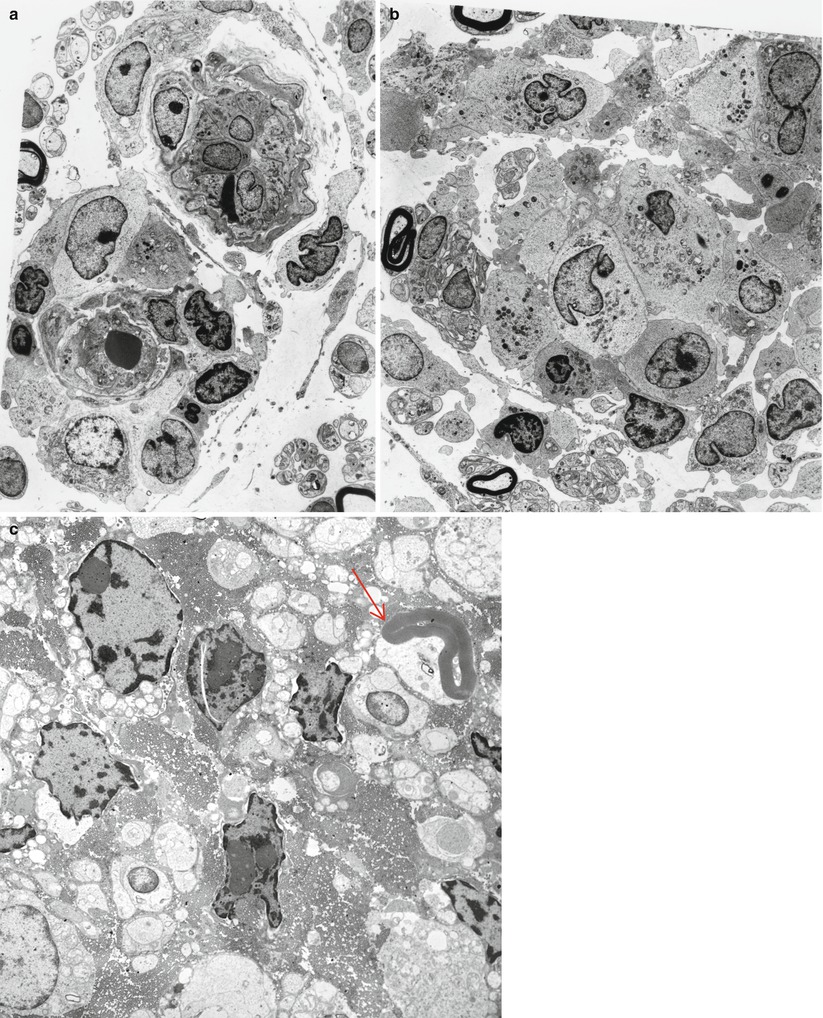
Most cases of SSN result from damage to the spinal dorsal root ganglia (DRG, Fig. 16.1a). Autopsy studies have shown a loss of DRG neurons and focal chronic inflammatory infiltrates, chiefly CD8-positive cytotoxic T cells (Wanschitz et al. 1997; Ichimura et al. 1998), with resultant degeneration of the dorsal columns (Horwich et al. 1977). Large neurons may be preferentially affected (Ohnishi and Ogawa 1986). These patients should not undergo sural nerve biopsy, as their syndrome is characteristic, biopsy pathology is nonspecific, and serological tests for the associated onconeural antibodies are available. However, diseases with characteristic histology which might mimic SSN include sarcoidosis, paraprotein-associated neuropathies, and CIDP.
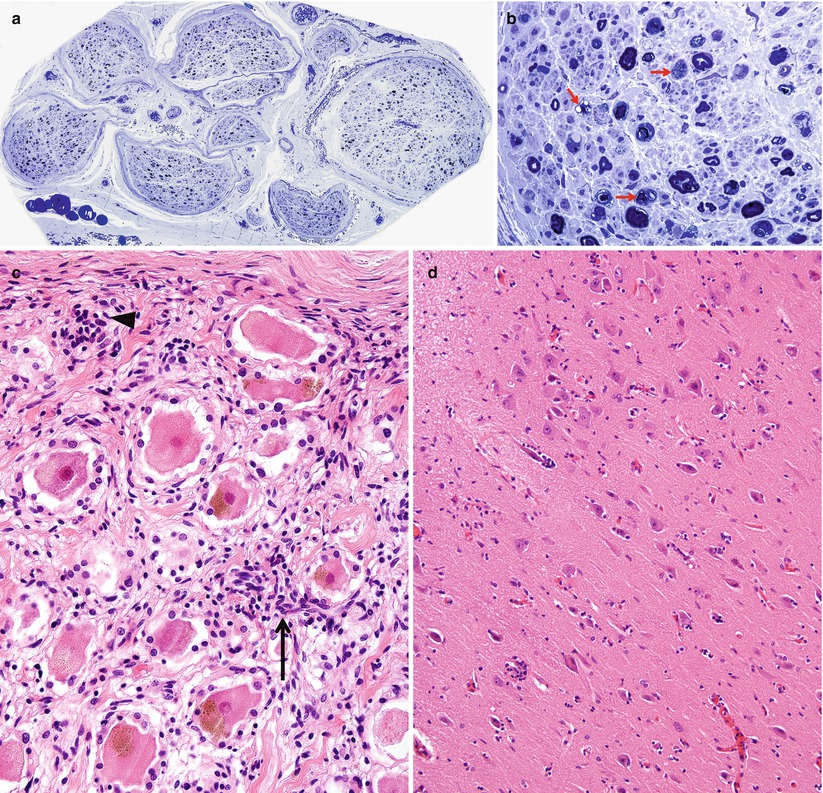
Fig. 16.1
Paraneoplastic subacute sensory neuropathy, radial nerve taken at the anatomical “snuffbox.” This 43-year-old woman was diagnosed as having small cell cancer of the lung following presentation with subacute sensory neuropathy involving the upper extremities. Note axon loss involving all fascicles (a) and marked active axonal degeneration affecting all fiber sizes (arrows, b). Regenerative axonal clusters are not seen. (c) Dorsal root ganglion of a different patient with small cell lung cancer and laboratory proven anti-Hu antibody. Inflammation at sites of neuron degeneration (arrow, c) and a developing residual nodule of Nageotte, a tombstone of prior neuronal degeneration (arrowhead, c) are shown. This patient also had limbic encephalitis with inflammatory changes in the hippocampus (d) (magnification: a, 40×; b, 1,000× 1 μ thick plastic sections; c 400×; d, 200×)
Nonetheless, pathological reports on a few patients are available (Lamarche and Vital 1987; Horwich et al. 1977; Dalmau et al. 1992; Dyck et al. 1958; Chalk et al. 1992). Sural nerve biopsy in patients with subacute sensory neuronopathy and marked sensory ataxia shows loss predominantly of large myelinated fibers, differing from preferential loss of small myelinated and unmyelinated axons in patients with pain predominant symptoms (Oki et al. 2007). Axon depletion in SSN ranges from mild to near total, with active degeneration a variably prominent feature (Fig. 16.1b, c). Axonal regenerative activity is usually minimal or absent, as expected in a neuronopathy (Lamarche and Vital 1987; Koike and Sobue 2008).
The majority of nerve biopsies reported in SSN have not shown inflammation, although one of our cases has shown significant sural inflammatory infiltration (Fig. 16.2a, b). However, prominent perivascular and vessel wall inflammation has occasionally been noted (Vallat et al. 1986; Plante-Bordeneuve et al. 1994) and, rarely, even vasculitis has been described (Johnson et al. 1979; Eggers et al. 1998). Such cases seem to reflect disease of the peripheral nerve rather than the spinal ganglion and may have a better prognosis in some patients (Plante-Bordeneuve et al. 1994) or may reflect a role for CV2/CRMP-5 which may occur with anti-Hu and be more targeted to peripheral nerve vs. dorsal root ganglia (vide infra).
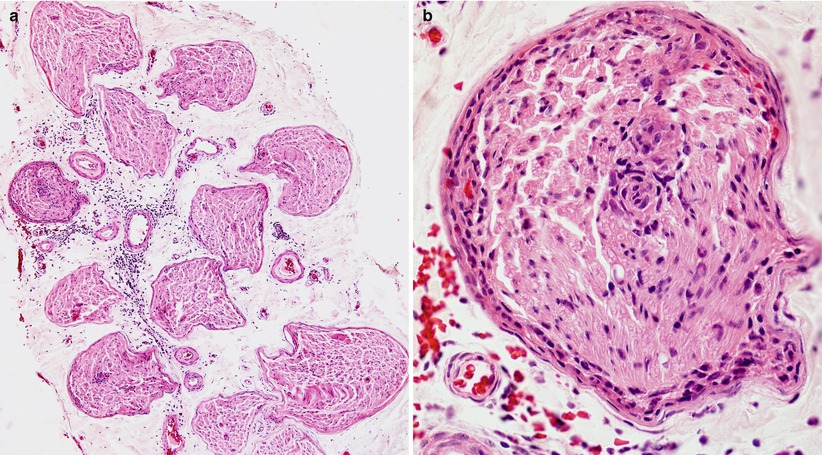
Fig. 16.2
Paraneoplastic neuropathy: sural nerve biopsy which led to the discovery of a small cell lung tumor in a 54-year-old woman. (a) Somewhat atypically, inflammation involves the epineurium, both perivascular and diffuse. (b) Higher magnification demonstrates prominent inflammatory infiltrate involving the perineurium and cuffing a small endoneurial vessel (paraffin, H&E, a, 100×; b, 600×)
16.1.1.3 Pathophysiology and Pathogenesis
The Paraneoplastic Neurological Syndrome Euronetwork (Giometto et al. 2010) has defined a number of antibodies (anti-Hu, Yo, CV2/CRMP-5, Ri, AChR, Ma2, Tr, and amphiphysin) which are thought to have a role in various paraneoplastic syndromes and may result in different clinical syndromes. Paraneoplastic SSN is most strongly associated with circulating antineuronal antibodies (anti-Hu, also called antineuronal nuclear antibody type 1, ANNA-1), which exhibits specificity for neuronal nuclear antigens (Smith 1992) and is most typically found in association with small cell lung cancer, much less frequently in other malignancies (Dalmau et al. 1992). It is thought that patients with anti-Hu antibodies alone most often present with subacute sensory neuronopathy, those with anti-CV2/CRMP-5 antibodies develop mixed axonal and demyelinating sensorimotor neuropathy, and those with both antibodies exhibit subacute sensory neuronopathy superimposed on demyelinating sensorimotor neuropathy (Antoine et al. 2001; Koike and Sobue 2013). Although patients with anti-Hu antibodies show a wide range of paraneoplastic neurological manifestations, including cerebellar ataxia, limbic encephalitis, myelopathy, Lambert–Eaton syndrome, and myopathy, peripheral neuropathy is the most common manifestation and present in 60–80 % of patients with SSN (Dalmau et al. 1992; Lucchinetti et al. 1998; Graus et al. 2001; Sillevis Smitt et al. 2002). The SSN DRG shown in Fig. 16.1c was accompanied by limbic encephalitis in the hippocampus (Fig. 16.1d). The anti-Hu antibody reacts with neoplasm antigens (Dalmau et al. 1992), leading to the assertion that the clinical syndrome is caused by antibodies produced against the tumor which cross-react with neuronal antigens. However, some of the lymphocytes infiltrating nervous tissue also appear to be sensitized to the Hu antigen, and cell-mediated damage may also play a role (Dalmau et al. 1992). In a previous study of 162 consecutive patients with anti-Hu antibodies, malignancy was present in 88 % of patients, and small cell lung cancer was subsequently identified in 93 % (Lucchinetti et al. 1998). CD8+ T cells in the DRG of SSN include both cytotoxic CD8+-positive T cells and atypical noncytotoxic CD8+-positive T cells with an uncertain dynamic (Roberts et al. 2009). It has been proposed that small cell lung cancer may evade surveillance by skewing tumor antigen-specific CD8+ T cells to an unusual noncytotoxic phenotype, providing an explanation for variation of clinicopathological features in anti-Hu antibody-associated paraneoplastic neurological syndromes (Roberts et al. 2009). Anti-CV2/CRMP-5 is also strongly associated with lung carcinoma (Honnorat et al. 1996; Yu et al. 2001), mostly small cell type, which was ultimately found in 77 % of patients with anti-CV2/CRMP-5 antibodies. Unlike anti-Hu antibodies, anti-CV2/CRMP-5 antibodies cross-react with antigens in the peripheral nerve trunk rather than DRG neurons (Antoine et al. 2001). Findings suggestive of demyelination and axonal degeneration have also been reported in biopsy specimens from patients with anti-CV2/CRMP-5 antibodies (Antoine et al. 2001). Somatic neuropathy is the most common manifestation in patients with anti-Hu and anti-CV2/CRMP-5 antibodies, while antiganglionic AchR antibody is associated with autonomic neuropathies (Graus et al. 2004). However, passive transfer of these antibodies or immunization has failed to reproduce the disease in animals (Sillevis Smitt et al. 1995).
A small proportion of patients with the antibody and the clinical syndrome have had no malignancy detected, even at autopsy (Dalmau et al. 1992). It is unclear whether the tumor was simply too small to be detected (Chartrand-Lefebvre et al. 1998; Lucchinetti et al. 1998) or the immune reaction to Hu antigen was caused by another factor. Fluorodeoxyglucose positron emission tomography (FDG-PET) may reveal the malignancy even if small (Rees et al. 2001; Younes-Mhenni et al. 2004; Titulaer et al. 2011).
16.1.2 Paraneoplastic Sensorimotor Neuropathy
16.1.2.1 Clinical Manifestations
Whereas SSN is a rare but characteristic paraneoplastic syndrome, a more nondescript sensorimotor neuropathy is seen in 5–16 % of cancer patients, with as many as 30–40 % showing subclinical electrophysiological abnormalities (McLeod 1993; Croft and Wilkinson 1965; Hawley et al. 1980; Walsh 1971; Graus et al. 1983; Paul et al. 1978). Small cell lung cancer is not as uniformly detected with these neuropathies as it is with SSN, and every type of malignancy has been associated with sensorimotor neuropathy, most often lung, breast, ovarian, stomach, and prostate cancers, as well as lymphoma (Croft and Wilkinson 1965; Walsh 1971; Dayan et al. 1965). Although the existence of this nonspecific sensorimotor “paraneoplastic” neuropathy is accepted, it is less precisely defined than SSN. Nonetheless, there is an association of patients with anti-CV2/CRMP-5 antibodies and the development of mixed axonal and demyelinating sensorimotor neuropathy, and those with anti-Hu and anti-CV2/CRMP-5 antibodies exhibit subacute sensory neuronopathy superimposed on demyelinating sensorimotor neuropathy (Antoine et al. 2001; Koike and Sobue 2013).
The paraneoplastic sensorimotor neuropathies are not homogenous, and classification is difficult (Corbo and Balmaceda 2001) because the pathophysiology of these entities is not understood and clinical features overlap. One approach that has been commonly used (Croft et al. 1967; McLeod 1993) distinguishes three groups on a clinical basis. Most frequently seen is a mild sensorimotor neuropathy that appears after the diagnosis of malignancy, sometimes called “mild terminal neuropathy”; the only abnormality may be hypoactivity of distal reflexes. The second group shows a more rapidly evolving and severe polyneuropathy that as often as not presents before the malignancy is diagnosed. The third and smallest group is composed of patients with a relapsing–remitting sensorimotor neuropathy. Regardless of where the patient falls into this classification, the neuropathic manifestations are typically sensorimotor, symmetrical, and predominantly distal, although proximal wasting and weakness can suggest a “neuromyopathy” (Croft and Wilkinson 1963). Motor neuron degeneration in the spinal cord at autopsy has been reported (Dalmau et al. 1992; Verma et al. 1996) and may be the cause of the motor component of paraneoplastic sensorimotor neuropathy in patients with anti-Hu-associated paraneoplastic neuropathy.
CSF protein is often elevated in the rapidly evolving and relapsing groups, but rarely in patients with “mild terminal neuropathy.” In this latter group, the electrophysiological assessment usually demonstrates axonal features, but in the more severe sensorimotor and the relapsing/remitting groups, a demyelinating or mixed pattern is often seen (Croft et al.1967; Paul et al. 1978; Graus et al. 1983).
As a rule, treatment of the tumor does little for the neuropathy, but exceptions have been reported, especially for lymphoma and seminoma (Evans and Kaufman 1971; Littler 1970; Enevoldson et al. 1990). Anecdotal reports indicate improvement with steroid or plasma exchange (Croft et al. 1967; Valbonesi et al. 1985).
16.1.2.2 Pathology
Autopsy studies have not shown disease of the spinal ganglia in paraneoplastic sensorimotor neuropathy except when SSN was superimposed (Croft et al. 1967). Inflammatory infiltrates have also been sparse in proximal nerve trunks (Croft et al. 1967). The literature on the peripheral nerve pathology of paraneoplastic neuropathy is scanty, since patients with a mild “terminal” neuropathy are unlikely to undergo a nerve biopsy. Moreover, some authors have not distinguished between the various sensorimotor neuropathy groups.
Mild Terminal Neuropathy
Sural nerve biopsy of patients with “mild terminal neuropathy” and lung cancer or lymphoma (Graus et al. 1983; Walsh 1971; Schlaepfer 1974) has revealed minimal reduction in total myelinated fiber density, but a significant decrease in large myelinated axons. At times active axonal degeneration and regenerating clusters have been present. Axons may appear thinly myelinated, and small onion bulbs can occasionally be seen. Schlaepfer (1974) suggested that the myelin alterations were secondary to axonal disease. Unmyelinated fibers are relatively or entirely spared (Walsh 1971). Inflammatory infiltrates are not a feature of “mild terminal neuropathy.” Immunofluorescence did not reveal immunoglobulin or complement deposition in the cases studied by Graus and co-workers (1983).
Severe Rapidly Evolving or Relapsing Sensorimotor Neuropathies
Croft and colleagues (1967) examined the peripheral nerve obtained at autopsy in 10 patients, 8 with subacutely evolving neuropathy and 2 with relapsing neuropathy. Slight lymphocytic infiltration was seen in 3 of 10 cases. Segmental demyelination and axonal degeneration were present, the former often predominating. Lamarche and Vital (1987) described nerve biopsies in six patients, most of whom had an acute or subacute neuropathy (Lamarche and Vital 1987). Axonal and demyelinating features were seen, the former usually predominating. Onion bulbs and regenerating clusters were often present. Inflammation was only identified in one case. Ultrastructural examination revealed that unmyelinated fibers were also affected, and uncompacted myelin was described in one instance. Graus et al. (1983) studied two patients with lymphoma and a severe neuropathy, one showing prominent demyelination and the other a mixed picture; inflammation was absent in both cases. The case of lymphoma reported by Sumi et al. (1983, case 1) showed typical features of macrophage-mediated demyelination and a course consistent with CIDP.
Although the demyelinating features of rapidly evolving paraneoplastic neuropathy have been emphasized in the literature, axonal neuropathies with no inflammatory features have also been described in this setting (Schlaepfer 1974; Enevoldson et al. 1990). In our experience, severe sensorimotor neuropathies associated with systemic malignancy may be axonal or demyelinating and inflammatory or noninflammatory. No specific features are present, and the diagnosis is made on clinical grounds.
Immunofluorescent studies have shown IgM and complement deposits in a subperineurial and perivascular localization (Ongerboer de Visser et al. 1983); however, this is a nonspecific finding.
Paraneoplastic “Microvasculitic” Neuropathy
A number of patients have been described who presented with a sensorimotor polyneuropathy in association with systemic malignancy and demonstrated “microvasculitis” on nerve biopsy (Vincent et al. 1986; Oh et al. 1991; Johnson et al. 1979; Younger et al. 1994). In the case reported by Younger et al. (1994), there was an associated anti-Hu antibody, but whether this was a causal factor or simply a coincidence is unknown. The descriptions and figures in these reports indicate an axonal neuropathy with intense perivascular cuffing and mononuclear infiltration of the vessel wall.
Use of the term “vasculitis” requires some evidence of vessel wall destruction, preferably fibrinoid necrosis and at the very least some evidence of disruption of the endothelium. While infiltration of the vessel wall is suggestive, it is not in itself sufficient to be considered vasculitis. Intramural lymphocytes may be in the process of migration into the endoneurium (Vallat et al. 1986). We too have seen axonal inflammatory neuropathies in which no etiology was found but a systemic malignancy was present, and we prefer to regard these as inflammatory neuropathies rather than “microvasculitis.” In lymphoid malignancies, nonspecific nonneoplastic inflammatory infiltrates seem to be a common finding (Dickenman and Chason 1958; Jellinger and Radiaszkiewicz 1976). The presence of prominent inflammation in “paraneoplastic neuropathy” may have therapeutic implications (Oh et al. 1991).
16.1.2.3 Pathogenesis
In contrast to paraneoplastic subacute sensory neuropathy, the cause of paraneoplastic sensorimotor neuropathy remains more obscure. Although previously no tumor-related immunologic alteration or toxin has been found to affect peripheral nerve (McLeod 1993; Ongerboer de Visser et al. 1983), more recent studies have identified onconeural antibodies. Specifically, patients with anti-CV2/CRMP-5 antibodies are now known to develop mixed axonal and demyelinating sensorimotor neuropathy, and those with both antibodies exhibit subacute sensory neuronopathy superimposed on demyelinating sensorimotor neuropathy (Antoine et al. 2001; Koike and Sobue 2013). In some studies, the frequency of neuropathy was correlated with weight loss (Hawley et al. 1980; Graus et al. 1983; Hildebrand and Coers 1967), but this is not consistent even for “mild terminal neuropathy” (Croft et al. 1967).
In the rapidly evolving or relapsing paraneoplastic neuropathies, segmental demyelination and onion bulbs have been seen, although not usually to the extent seen in CIDP or other hypertrophic neuropathies. This suggests an important role for myelin injury. Indeed, CIDP may be “precipitated” by immunologic disturbances associated with the tumor (Sumi et al. 1983). However, segmental demyelination in paraneoplastic sensorimotor neuropathy may be secondary, as suggested by some teased fiber studies (Schlaepfer 1974; Lamarche and Vital 1987; Walsh 1971).
Some paraneoplastic sensorimotor neuropathies may be “dying-back” axonopathies. Intramuscular nerve axonal swelling has been described as paraneoplastic phenomenon by several authors (Barron and Heffner 1978; Brownell and Hughes 1975; Hildebrand and Coers 1967; Awad 1968) and may represent the earliest stages of a distal axonopathy as seen first in the longest motor fibers (Barron and Heffner 1978); similar changes are seen in typical toxic distal axonopathies (Spencer and Schaumburg 1977). However, the specificity and significance of this finding are suspect given its high frequency in normal intramuscular nerves and in neuropathies which are not believed to be due to a “dying-back” process (Alderson 1992).
16.1.3 Other Associations Between Neuropathy and Malignancy
16.1.3.1 Necrotizing Peripheral Nerve Vasculitis
The co-occurrence of vasculitis and malignancy is well recognized (Sanchez-Guerrero et al. 1990), but peripheral nerve involvement seems to be rare. Nonetheless, peripheral nerve necrotizing vasculitis has been described in association with malignancy. Patients may present with a sensorimotor polyneuropathy (Harati and Niakan 1986; Torvik and Berntzen 1968; Naka et al. 1991) or as pure sensory syndrome (Johnson et al. 1979). Deposition of immunoglobulin or complement in the vessel has not been detected (Johnson et al. 1979). Given the infrequency of this association, the possibility of a chance co-occurrence of peripheral nerve vasculitis and systemic malignancy must be considered.
16.1.3.2 Acute Demyelinating Neuropathy
Guillain–Barré syndrome has been infrequently reported in association with Hodgkin’s disease (Lisak et al. 1977; Ropper et al. 1991; Julien et al. 1980; Vital et al. 1990). No special features characterized the few cases studied pathologically, and typical macrophage-mediated demyelination has been observed (Julien et al. 1980).
16.1.4 Differential Diagnosis
Identification of paraneoplastic neuropathy may lead to early detection of malignancy. A paraneoplastic basis should be considered in any sensorimotor axonal or mixed neuropathy, inflammatory or not, when no other etiology can be determined. As lung cancer and lymphoma account for a large fraction of the associated malignancies, a thorough physical exam and a chest X-ray would go a long way toward excluding the possibility of an occult malignancy. Unfortunately, nerve biopsy does not reveal any characteristic histological features to suggest the diagnosis and should only be performed when an alternative and treatable diagnosis is under consideration.
16.2 Neuropathy Due to Neoplastic Infiltration
Diffuse infiltration of nerve with malignant cells is an uncommon cause of neuropathy. This is essentially confined to hematologic malignancy, including lymphoma, leukemia, myeloma, and a few other rare entities. Solid organ cancers, such as those of the ear, nose, and throat (ENT), pancreas, and prostate, often produce peripheral nerve dysfunction by compression or gross infiltration of roots, plexus, or focal segments of nerve but do not produce a diffuse infiltrative neuropathy (Grisold et al. 2013) and are thus not considered.
16.2.1 Lymphomatous Neuropathy
16.2.1.1 Clinical Manifestations
Lymphomatous neuropathy is diagnosed when malignant lymphoma infiltrates and damages the peripheral nerve. This has been called neurolymphomatosis by some (Diaz-Arrastia et al. 1992), although others have taken nosologic exception (Guberman 1984; Baehring et al. 2003; Grisariu et al. 2010). Diaz-Arrastia et al. (1992) have recently reviewed the subject and examined a total of forty cases from the literature. Clearly the disorder is much more common than such numbers would indicate, as we have seen 4 cases in our own laboratory. About half of patients are not known to have systemic lymphoma at the time of diagnosis. Even at autopsy, 7 of 22 patients had lymphoma restricted to the nervous system alone, 2 had a focal lymphoma with dissemination to peripheral nerves only, and disseminated lymphoma was present in 13 (Diaz-Arrastia et al. 1992). The lymphoma is almost invariably of the non-Hodgkin (NHL) type (98 %), and 75 % of appropriately studied cases were of the B-cell malignancies (Diaz-Arrastia et al. 1992). Neuropathy is usually diffuse and sensorimotor in type and can evolve acutely mimicking Guillain–Barré syndrome, in a relapsing–remitting fashion, or most often progressing relentlessly over weeks to months (Diaz-Arrastia et al. 1992). One of the cases we have studied progressed slowly over 2 years. Pain may be a prominent feature. Electrophysiological testing can demonstrate axonal, demyelinating, or mixed features, and CSF examination often reveals elevated protein or CSF pleocytosis, with malignant cells in about half of cases (Diaz-Arrastia et al. 1992). Steroids, chemotherapy, or radiation therapy may be helpful. While the prognosis is usually poor (Diaz-Arrastia et al. 1992), in some patients, appropriate treatment may result in survival for many years (Ince et al. 1987).
Also intravascular lymphoma, formerly named “Tappeiner” disease or neoplastic angioendotheliomatosis, has been added to the concept of lymphomatous involvement (Glass et al. 1993). It can present as multiplex neuropathy, as Guillain–Barré syndrome (GBS) (Jiang et al. 2010), and as chronic inflammatory demyelinating polyneuropathy (CIDP) (Grisold et al. 2007; Briani et al. 2009, 2011). Intravascular lymphoma (Glass et al. 1993) is typically of B-cell origin and is thought to reflect lack of CD29 (β-1 integrin) and CD54 (ICAM-1) adhesion molecules on the neoplastic lymphoma cells interfering with transvascular migration (Ponzoni et al. 2000). Cauda equina syndrome, ascending polyradiculopathies (Levin and Lutz 1996; Patel et al. 2006; Jiang et al. 2010), multiple mononeuropathies (Roux et al. 1995), isolated mononeuropathies (Vital et al. 1989), symmetrical neuropathies (Oei et al. 2002), and muscle involvement (Fallon et al. 2002) have been observed (reviewed in Grisold et al. 2013).
16.2.1.2 Pathology
Because only half of patients are known to have systemic lymphoma, neurolymphomatosis is one instance where nerve biopsy may be essential in making the diagnosis (Diaz-Arrastia et al. 1992). Even if malignancy is known to be present, infiltration of the nerve may be the only visible manifestation of recurrence after treatment (Shoenfelt et al. 1983; Krendel et al. 1991). Descriptions below are based on literature material (Barron et al. 1960, case 5; Gherardi et al. 1986; Guberman et al. 1978; Ince et al. 1987; Krendel et al. 1991; Thomas et al. 1990; Vital et al. 1990; Diaz-Arrastia et al. 1992; Zuber et al. 1987; Kohut 1946; Stack 1991; Atiq et al. 1992; Shoenfeld et al. 1983; Gold et al. 1988) and our experience with 4 cases – two at autopsy and two at nerve biopsy.
Autopsy reviews provide an inconsistent picture of the frequency of peripheral nerve involvement. In two autopsy series of Hodgkin’s disease, peripheral nerve tumor infiltration was detected in 0/30 and 5/30 patients, but far more often there were nonspecific nonneoplastic inflammatory infiltrates (Dickenman and Chason 1958; Jellinger and Radiaszkiewicz 1976). In the study of Grisariu et al. (2010), neurolymphomatosis was due to non-Hodgkin’s lymphoma in 90 % of cases and to acute leukemia in 10 %. In up to 26 % of the cases, it occurred as an initial manifestation of malignancy. We have found only one report of Hodgkin’s lymphoma causing a histologically proven infiltrative clinical neuropathy (Barron et al. 1960). In NHL the incidence of detected peripheral nerve infiltration by tumor cells in autopsy material varied from 0/20 to 41/98 (Dickenman and Chason 1958; Jellinger and Radiaszkiewicz 1976), but the quantity and location of peripheral nervous tissue studied was not clearly specified. A wide variety of NHL types have been present, including cutaneous and non-cutaneous T-cell lymphomas and high- and low-grade B-cell lymphomas. No particular subtype emerges as particularly likely to infiltrate peripheral nerve. Analysis of the literature is limited by small numbers and lack of the modern immunohistochemical and cytological techniques now routinely used to categorize lymphoma.
Light Microscopy
In our experience, a diffuse massive infiltration of all peripheral nerve compartments is most typical of lymphoma, although fascicles may be differentially involved (Figs. 16.3 and 16.4). The infiltrate may demonstrate epineurial predominance or prominent perineurial and subperineurial localization (Fig. 16.3). Selective exclusive endoneurial involvement is rare and has been described after treatment with chemotherapy (Schoenfeld 1983; Krendel et al. 1991), but may also occur in untreated patients (Atiq et al. 1992). The illustrated case of neurolymphomatosis showed small epineurial perivascular foci of atypical cells in only 2 of 38 tissue blocks (Fig. 16.4b). This underscores the importance of examining the maximum amount of tissue possible.
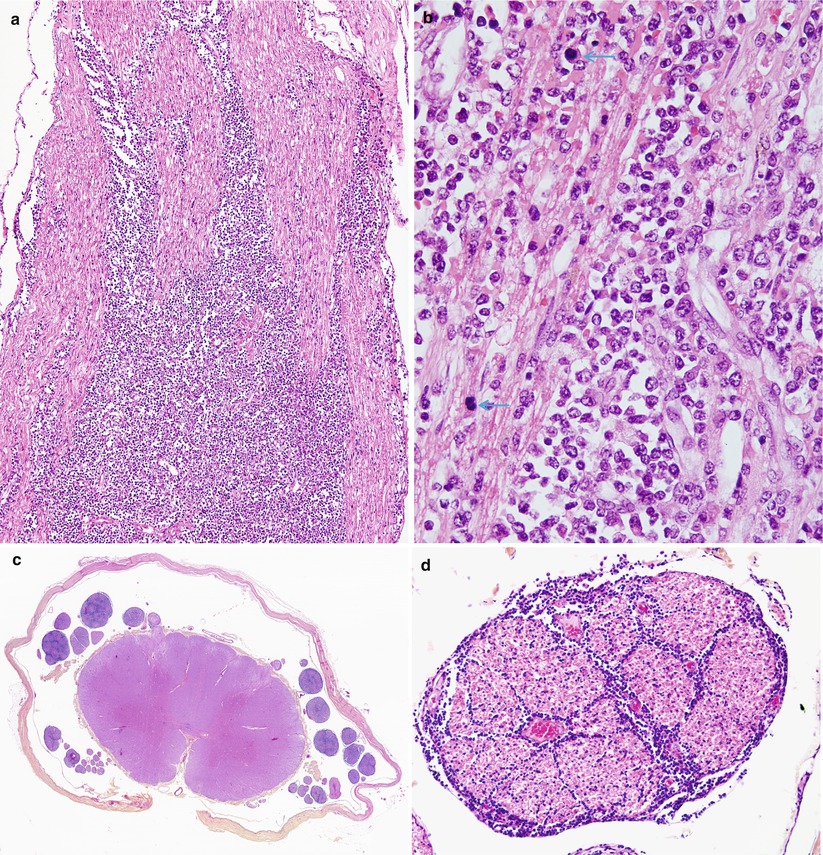
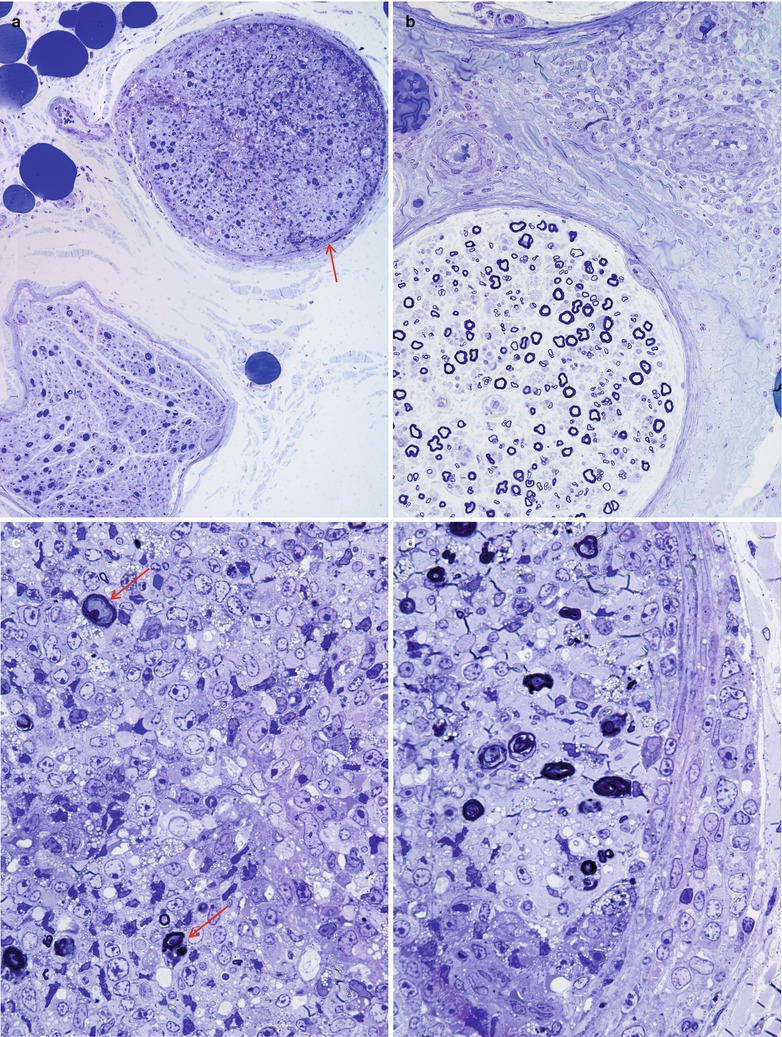
Fig. 16.3
Neurolymphomatosis: longitudinal view demonstrates streaming of lymphomatous infiltrate between axons (a). The presence of markedly atypical cells and numerous mitoses (arrows) indicates a high-grade lymphoma. In addition to sural nerve involvement, there was extensive infiltration of the dorsal and ventral roots (c) with extension into the perineurium and endoneurium (d) (paraffin)
Fig. 16.4
Neurolymphomatosis: (a) lymphomatous infiltration involves one fascicle (arrow) of sural nerve but spares another. (b) Atypical perivascular mononuclear infiltrate in the epineurium. (c) Higher magnification of the involved fascicle shows replacement of the endoneurial contents by lymphoma sparing a few myelinated axons (arrows). (d) Part of the perineurium is disrupted by neoplastic lymphocytes (paraffin, magnification a, 200×; b, 400×; 1 μ thick plastic sections c, d, 1,000×)
A tendency toward perivascular cuffing is commonly seen, and sometimes a striking angiocentricity of the tumor cells is present. The infiltrating cells may be found within vessel walls but do not cause vascular necrosis. Reticulin stains will demonstrate the formation of a delicate network around vessels and within collections of tumor cells. Proliferation of epineurial or endoneurial vessels may be seen (Gherardi et al. 1986; Zuber et al. 1987). Mitotic figures, pleomorphism, and atypia of the infiltrating cells are usually immediately suggestive of the diagnosis (Figs. 16.3, 16.4 and 16.5). However, well-differentiated lymphoma cells might be difficult to distinguish from mature lymphocytes (Fig. 16.5).