and Robert E. Schmidt2
(1)
Sunnybrook and St Michael’s Hospitals, University of Toronto, Toronto, ON, Canada
(2)
Division of Neuropathology Department of Pathology, Washington University School of Medicine, St. Louis, MO, USA
Inborn errors of metabolism can manifest as multisystem diseases characterized by abnormal intracellular accumulation of storage material, the best known involving lysosomal enzymes. In a recent review (Boustany 2013), a less restrictive classification of lysosomal storage disorders has been proposed which includes diseases that display defects in cellular storage, synthetic enzymes, lysosome membrane or other membrane proteins, and trafficking. In the past, tissue examination for identification and characterization of storage material, including nerve biopsy, was an important means of diagnosis. However, advances in understanding of the biochemical and genetic basis of disease have revealed specific enzymatic and genetic defects in many of these conditions (Table 20.1), and noninvasive diagnosis using genetic techniques, assays of enzyme activity in fluids or cultured cells, or detection of abnormal storage products has increasingly become the diagnostic method of choice. Nevertheless, atypical clinical manifestations or inaccessibility of advanced biochemical and genetic techniques occasionally make nerve biopsy useful in some patients, and for a small number of storage diseases, histologic examination remains essential.
Table 20.1
Storage diseases causing polyneuropathy
Sphingolipidoses | Protein/enzyme defect |
Metachromatic leukodystrophies | Arylsulfatase A (cerebroside sulfatase) |
Saposin B arylsulfatase A activator | |
Arylsulfatase A, B, and C (multiple sulfatase deficiency) | |
Globoid cell leukodystrophy (Krabbe disease) | Galactosylceramide-β-galactosidase |
Fabry disease (angiokeratoma corporis diffusum) | α-Galactosidase A |
Niemann–Pick disease (type A) | Acid sphingomyelinase |
Farber disease (lipogranulomatosis) | Acid ceramidase |
GM1 gangliosidosis | β-Galactosidase |
GM2 gangliosidosis (various syndromes) | Hexosaminidase A |
GM2-activator protein | |
Others | |
Adrenoleukodystrophy (X-linked form) | ABCD1 peroxisomal transporter |
Tangier disease | Alpha lipoprotein |
Cerebrotendinous xanthomatosis | Mitochondrial sterol-27-hydroxylase |
Glycogenosis type III (Cori–Forbes) | Debrancher enzyme (amylo-1,6-glucosidase) |
Sialidosis type I | Glycoprotein specific α-neuraminidase |
Wolman disease | Lysosomal acid lipase/cholesteryl esterase |
Niemann-Pick disease (type C) | NPC1 gene related product |
20.1 The Sphingolipidoses
Sphingolipids are lipids in which sphingosine, a complex amino alcohol, replaces glycerol in the lipid backbone. This class of molecules includes gangliosides, sulfatides, and cerebrosides and plays a role in biological membrane structure and function. Specific enzymatic defects result in pathological accumulation of lipid within lysosomes in various tissues, although how this leads to cell injury is often not known. The diseases included under this aegis include metachromatic leukodystrophy, globoid cell leukodystrophy (Krabbe disease), Fabry disease, Niemann–Pick disease, Farber disease, GM1 and GM2 gangliosidosis, and Gaucher disease.
20.1.1 The Metachromatic Leukodystrophies
The metachromatic leukodystrophies (MLD) are caused by defective activity of the arylsulfatase enzyme system, with a consequent accumulation of sulfatides. The most common variant (Phenotype MIM#250100) is caused by a defect in the arylsulfatase A gene (ARSA, protein product cerebroside sulfatase). The AB variant of MLD (MIM#249900) is produced by a mutation in the prosaposin gene (PSAP) on chromosome 10q22.1 which produces a deficiency of sphingolipid activator protein saposin B resulting in diminished ARSA activity. Multiple sulfatase deficiency (MIM#272200) is produced by a defect of arylsulfatases A, B, and C due to mutation in the sulfatase–modifying factor–1 gene (SUMF1). This form is clinically similar to the others but includes the presence of urinary mucopolysaccharides in addition to sulfatides (Hahn et al. 1981; Thomas 1993; Fluharty 2006). All are transmitted in autosomal recessive fashion. Finally, ARSA pseudodeficiency (MIM#250100), in which ARSA enzyme activity is 5–20 % that of normal controls, has been reported in otherwise healthy individuals. The clinical picture of MLD has been divided into infantile (50–60 % cases, juvenile (20–30 %), and adult-onset variants (15–20 %, Fluharty 2006), with the age of onset being consistent within a pedigree (Alves et al. 1986; Haltia et al. 1980; Percy et al. 1977). Advances in molecular biology have validated these subdivisions (Polten et al. 1991).
20.1.1.1 Clinical Manifestations
Infantile cases present before the third year of life with motor delay or regression and cognitive difficulties. As the disease progresses, ataxia and hypotonia appear and mental function worsens, with death resulting within the first decade. The adult form of ARSA deficiency, defined as age of onset after 15–21 years of life, is much less common than the infantile variety and usually presents as a disturbance of mental function with subsequent evidence of diffuse CNS disease, including visual loss, ataxia, and movement disorders. Severe debilitation and death are inevitable, but may take 15 or more years to occur. Manifestations of the juvenile form are intermediate between those of the infantile and adult variants. Multiple sulfatase deficiency is clinically more severe than ARSA deficiency, with organomegaly and ichthyosis being unique to this variant (Thomas 1993).
While not ordinarily the dominant manifestation of MLD in any of its variant forms, peripheral neuropathy is usually present, albeit sometimes only electrophysiologically (Percy et al. 1977; Vos et al. 1982). Rare reports document a chronic progressive demyelinating neuropathy as the presenting manifestation of MLD in adults or children (Fressinaud et al. 1992; Vos et al. 1982; Yudell et al. 1967). The demyelinating nature of the neuropathy is evidenced by a marked slowing of conduction velocities (Vos et al. 1982; Martin et al. 1982a).
Diagnosis of MLD is usually easily made by demonstration of decreased cerebroside sulfatase activity in serum samples or cultured cells or by analysis of a gene defect. However, enzyme assays are normal in the rare cases of defective arylsulfatase A activator protein (Hahn et al. 1981; Shapiro et al. 1979; Stevens et al. 1981). Conversely, a relatively common pseudodeficiency of ARSA has been identified which can make assays of enzyme activity misleading (Hohenschutz et al. 1989; Kappler et al. 1991; Penzien et al. 1993). Without biochemical testing of family members, interpretation of low ASA activity can be difficult (Vos et al. 1982). Accumulation of sulfatides separates MLD from ARSA pseudodeficiency, is seen in all MLD variants, and is traditionally demonstrated by the finding of metachromasia in urinary sediment cells or a quantitative increase in urinary sulfatide excretion (Moser 1985). Identification of defects in arylsulfatase A ARSA, prosaposin (PSAP), or sulfatase-modifying factor–1 gene (SUMF1) now obviates some of the confusion.
20.1.1.2 Pathology
The central nervous system shows diffuse demyelination sparing subcortical “U” fibers, with some loss of neurons, and numerous metachromatic-debris-filled macrophages (Gregoire et al. 1966; Haberland et al. 1973; Alves et al. 1986; Satoh et al. 1988). The ultrastructure of the inclusions seen in the CNS is similar to that of the PNS as described below (Gregoire et al. 1966). Metachromatic storage material is also detected in non-nervous tissues (Haberland et al. 1973; Gregoire et al. 1966; Satoh et al. 1988).
Role of Biopsy
The literature on nerve biopsy in MLD is considerable (Vos et al. 1982; Martin et al. 1982a; Thomas et al. 1977; Percy et al. 1977; Bardosi et al. 1987; Bischoff 1979). Characteristic storage material can probably be seen in all nerve biopsies postnatally, even well before the development of clinical and electrophysiological abnormalities (Argyrakis et al. 1977). Storage substance was reported at 23 and 26 weeks of gestation, even before the appearance of demyelinating changes (Martin et al. 1982a; Meier and Bischoff 1976). In one report of two siblings who had no characteristic inclusions in nerve despite clinical and electrophysiological evidence of a neuropathy, the diagnosis of MLD is questionable because of very atypical biochemical features (Iannaccone et al. 1993). Skin biopsy is less likely to be helpful (Dolman et al. 1975).
Nerve biopsy is usually not required for the diagnosis of MLD. The diagnosis of MLD must be confirmed by one or more of the following additional tests: molecular genetic testing of ARSA (the only gene in which mutation is known to cause arylsulfatase A deficiency), urinary excretion of sulfatides, and/or finding of metachromatic lipid deposits in nervous system tissue (Fluharty 2006). However, when biochemical tests are falsely negative, biopsy may be of value. MLD may not be suspected in those rare patients who have a neuropathy without detectable CNS disease, and nerve biopsy will lead to a surprise diagnosis (Fressinaud et al. 1992; Vos et al. 1982, case 20). In a report by Vos and colleagues (1982) the authors found nerve biopsy exposed misleading biochemical information in 2 of 22 patients, resulted in an unexpected diagnosis of MLD in 1 patient, and helped distinguish between MLD and a competing diagnosis in 2 further patients.
Light Microscopy
Histochemical properties of the storage material of MLD are reliably assessed only on frozen material. A search for brown or brown-yellow metachromasia with acidified cresyl violet (Hirsch–Peiffer technique) or toluidine blue on frozen sections appears to be the method of choice, although other aniline stains such as acridine or toluidine blue will also reveal metachromasia (Olsson and Sourander 1969). The storage material of MLD is also PAS positive, and with cresyl-violet staining, polarized light demonstrates yellow-green dichroism (Dayan 1967; Takahashi and Naito 1985). Sections stained with acriflavine may demonstrate a yellow-orange secondary fluorescence (Olsson and Sourander 1969).
The staining properties of Reich Pi granules on frozen sections are similar to those of MLD inclusions (Olsson and Sourander 1969), although Dayan (1967) indicated that Pi granules stained red, not brown, with the Hirsch–Peiffer technique. The perinuclear and paranodal “storage material” observed by Dayan (1967) in teased fibers of a 15-year-old with MLD might have represented Pi granules, but, if so, the quantity of this material would have been most remarkable for this age. Prior to the availability of electron microscopy, the diagnosis of MLD could be made when the metachromatic material was present in large quantities and especially when it was seen in endoneurial phagocytes; subtle Schwannian accumulation of metachromatic material could not be reliably interpreted (Olsson and Sourander 1969). Electron microscopy is highly sensitive in detecting inclusion material and demonstrates a variety of pathognomonic ultrastructural appearances.
In Schwann cells, storage material is concentrated most densely in the perinuclear region and takes the form of 0.5–2.0 μm granules. In addition, myelinating Schwann cells often demonstrate, external to the myelin sheath, concentric lamellar osmiophilic bodies surrounded by a halo which do not represent metachromatic storage material (vide infra). Endoneurial macrophages, usually arrayed in a perivascular distribution, can contain inclusion material.
In MLD, the myelinated axon population is often reduced, and axons of all sizes are lost such that the diameter–frequency histogram may retain its bimodal appearance (Martin et al. 1982a; Bardosi et al. 1987). Actively degenerating axons and regenerating clusters are uncommon. Unmyelinated fibers have been less rigorously studied, but seem to be spared (Case Records NEJM 1984). A picture of diffuse hypomyelination is usually present (Fig. 20.1a, b), and small onion bulb formations may be seen, in both infantile and adult cases (Bardosi et al. 1987; Martin et al. 1982a; Thomas et al. 1977; Vos et al. 1982). The onion bulbs are usually rudimentary, but were said to be quite prominent in the five cases of juvenile MLD reported by Haltia et al. (1980) and in a report of MLD due to activator protein deficiency (Hahn et al. 1981). The myelin sheath is often irregular, with evidence of remodeling and occasional formation of redundant myelin loops. Active demyelination, with naked axons and debris-filled macrophages (Fig. 20.2), is seen most prominently in the infantile cases, the macrophages often congregating in a perivascular or subperineurial distribution. In the adult cases the process is usually more indolent and active demyelination and debris-filled cells (Fig. 20.2) are uncommon (Martin et al. 1982a; Thomas et al. 1977; Haltia et al. 1980). However, Percy and colleagues (1977) found that the pace of demyelination correlated more with the disease stage than the age of onset, i.e., a biopsy taken during the rapidly progressive early stage of adult-onset disease showed the same active demyelination as that of an infantile-onset case. Fibrotic changes may be prominent.
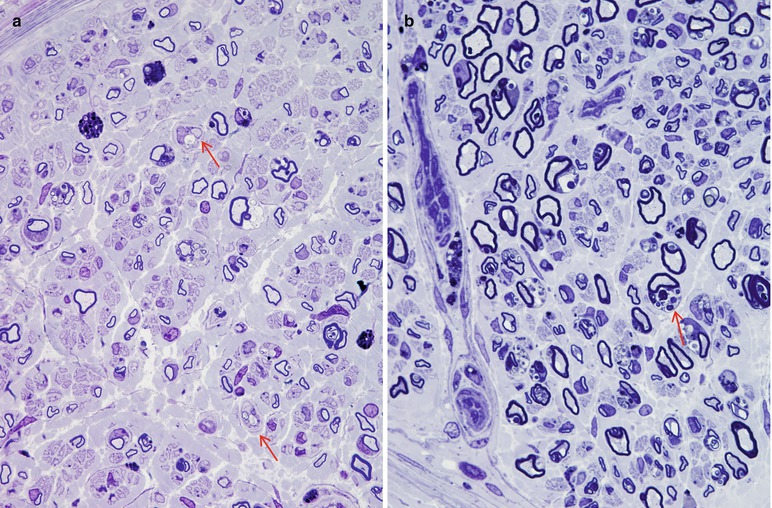
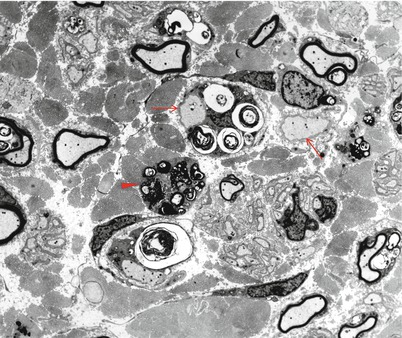
Fig. 20.1
MLD: most fibers are thinly myelinated (a, b). Segmental demyelination is seen in (a) (arrows), and many Schwann cells contain concentric lamellar osmiophilic bodies (arrow, b) (1 μm thick plastic sections: a, 600×; b, 1,000×) (Tissue courtesy of Dr. D. Agamanolis)
Fig. 20.2
MLD: low magnification view of the sural nerve shows constituent elements ranging from naked axons (arrows) to intact, thinly myelinated elements and macrophages containing phagocytosed sulfatide (arrowhead) (2,500×)
Studies of teased fibers have shown that segmental demyelination is the predominant pathological process and that this does not correlate with the local prominence of inclusions (Dayan 1967).
Electron Microscopy
The storage material of MLD is single membrane bound and can take a variety of ultrastructural appearances. “Tuffstone” bodies are readily identified as sharply demarcated inclusions containing an intermingling of granular material, electron-lucent vacuoles, and regions with periodicity (Figs. 20.3a, b and 20.4a, b). A second type of inclusion ultrastructure (Fig. 20.5a, b) has been described under a confusing plethora of names including “stacked disc,” “prismatic,” or “herringbone” inclusions (Gregoire et al. 1966; Luijten et al. 1978; Martin et al. 1982a; Rutsaert et al. 1973). Depending on the plane of section, the appearance may range from maze-like or honeycombed to parallel arrays of lamellated material, and we will use the term “prismatic” to describe these various morphologies, as it seems to have been applied most consistently in the literature. Inclusions best categorized as “tuffstone bodies” often show regions of prismatic structure which blend into less structured material, suggesting that there is an evolution from one appearance of the stored material into the other. A third type of storage cytosome is the “zebra body,” an oval or round membrane bound structure traversed by alternating light and dark bands, the latter made up of several closely packed lamellae. When seen in myelinating Schwann cells, these may be confused with Reich Pi granules, but this is less of an issue when detected in nonmyelinating Schwann cells and macrophages (Bischoff 1979). The various inclusion morphologies blend into each other and thus probably reflect different orientations and packing of the same storage material. Regardless of inclusion type, periodicity of storage material, when present, is 5.6–6.0 nm, visibly less than that of myelin, and typical of the appearance of stored intracellular sulfatide in experimental work (Rutsaert et al. 1973).
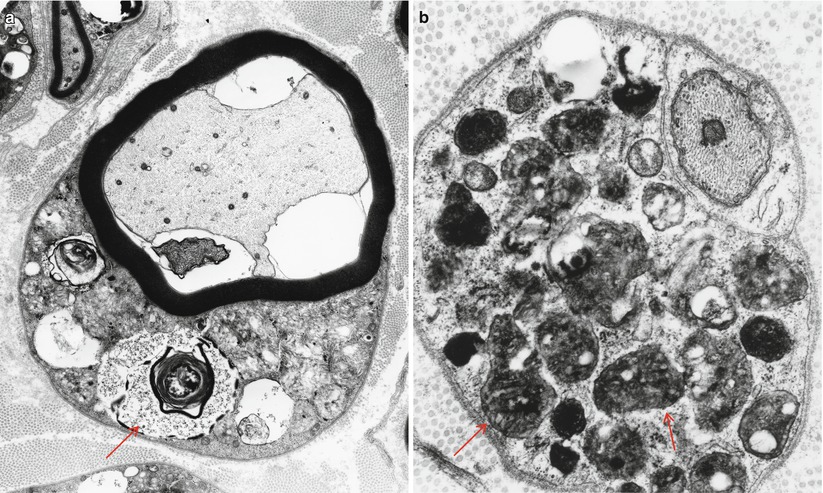
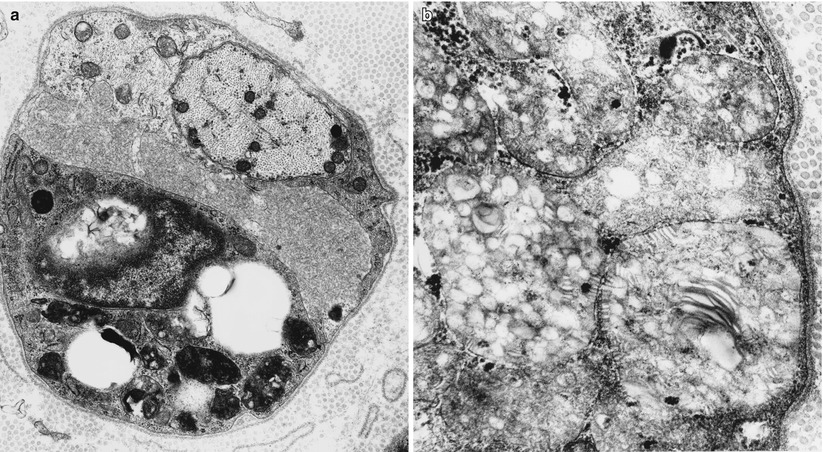
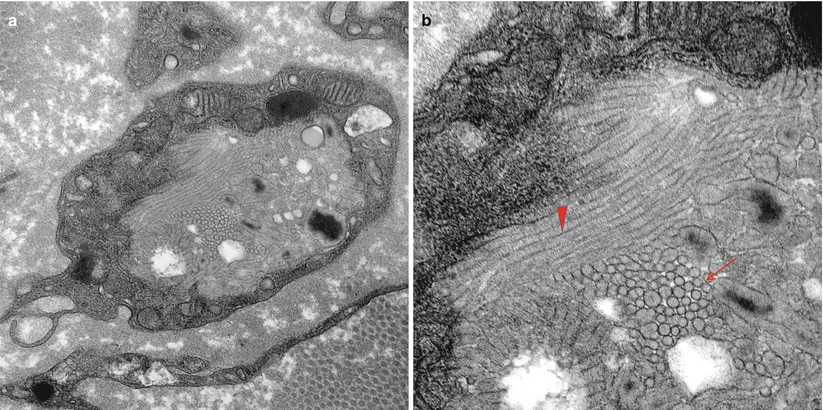
Fig. 20.3
MLD: (a) EM reveals that the concentric lamellar structures seen on LM are secondary lysosomes containing a myeloid whorl surrounded by glycogen (arrow, a). (b) In comparison, the characteristic inclusion of MLD, “Tuffstone” bodies (arrows) are seen (a, 11,000×; b, 23,660×)
Fig. 20.4
“Tuffstone” inclusions are seen within the Schwann cell cytoplasm surrounding a demyelinated axon and at higher magnification (b) (a, 13,520×; b, 49,010×)
Fig. 20.5
(a, b) Large prismatic inclusion in macrophage seen in stacked (arrow, b) and longitudinal orientation (arrowhead, b) (a, 25,000×; b, 60,000×)
The storage material is seen in myelinating Schwann cells, nonmyelinating Schwann cells (and, thus, are not derived from myelin breakdown), and in macrophages. Myelinating Schwann cells are most likely to contain tuffstone bodies, while macrophages most often show “prismatic” inclusions (Martin et al. 1982a), both likely derived from lysosomes. The single detailed histological report of MLD with activator deficiency demonstrated typical findings, but also revealed lamellated material in endothelial cells (Hahn et al. 1981). Although not mentioned in most reports, a few authors have stated that inclusions were observed in axons, and one example has been illustrated (Thomas et al. 1977).
Inclusions are seen regardless of the age of onset, although they are more numerous in the infantile cases (20–45 % of myelinated fibers) than in adult- or juvenile-onset cases (10 % of myelinated fibers) (Martin et al. 1982a). Luijten et al. (1978) felt that ultrastructure of the inclusions varied with age, infantile cases characterized by tuffstone bodies, juvenile cases showing prismatic inclusions, and adult patients mostly displaying zebra bodies, but this was based on the study of only one case in each group. The ten patients studied by Martin et al. (1982a) showed a trend towards this pattern, but overall, the full spectrum of ultrastructural pathology has been seen regardless of age of onset and genetic subtype (Hahn et al. 1981; Martin et al. 1982a; Thomas et al. 1977).
In MLD, structures composed of concentric lamellar osmiophilic substance are often seen in myelinated fibers, usually external to an otherwise normally compacted sheath; these correspond to the ovoids seen on LM (Cravioto et al. 1966). The difference in periodicity between these structures and the inclusions of MLD, the observation that the periodicity is similar to myelin (Argyrakis et al. 1977), and the occasional detection of continuity between the myeloid whorl and intact compact myelin (Cravioto et al. 1966) suggest that this structure is a stage in the remodeling of the myelin sheath and not directly related to the storage material. Indeed, similar myelin configurations are seen in a wide variety of neuropathies, albeit not usually so prominently. However, Thomas et al. (1977) found the lamellated bodies to have a periodicity of 8 nm and failed to detect sites of continuity with intact myelin. Thus, an alternative possibility is that these bodies represent appearance of lipid storage material being degraded within secondary lysosomes. Cellular organelles may show nonspecific changes such as increases in glycogen (Argyrakis et al. 1977), abnormally formed mitochondria (Argyrakis et al. 1977; Cravioto et al. 1966), or dilation of the endoplasmic reticulum (Argyrakis et al. 1977).
A peculiar “loosened” myelin appearance has been illustrated in MLD on several occasions (Cravioto et al. 1966; Percy et al. 1977; Hahn et al. 1981; Webster 1962; Bischoff 1979). This differs from the widely spaced myelin seen in some paraprotein-associated neuropathies (Chap. 14) in that “loose” myelin is often wavy and the separation between major dense lines is less uniform. Similar “loosened” myelin sheaths are illustrated by Bischoff (1979) in diabetic and uremic neuropathies (Figs. 4.38, 4.39 reference Bischoff 1979) and by Vital and Vallat in idiopathic and leprous neuropathies (Figs. 90, 91, 168, 169 in Vital and Vallat 1987). We have never observed this alteration and wonder whether it represents an artifact of fixation or tissue trauma to which nerves in MLD patients are more susceptible.
20.1.1.3 Pathogenesis and Therapy
The common denominator of all the MLD is the accumulation of sulfatide, and this has been shown to be the major component of the material accumulated in the characteristic metachromatic inclusions (Fig. 20.6) (Suzuki et al. 1967). The storage material is seen prior to the onset of myelination and in nonmyelinating Schwann cells and is thus unlikely to reflect uncleared myelin debris (Meier and Bischoff 1976). The lack of correlation between sulfatide accumulation and the presence of segmental myelin damage (Martin et al. 1982a; Dayan 1967) suggests that Schwann cell dysfunction is not necessarily due to toxic accumulations of storage material. An alteration in the fatty acid composition and possibly the physical properties of myelin may be present in MLD, and this may somehow result in myelin damage (Thomas 1993).
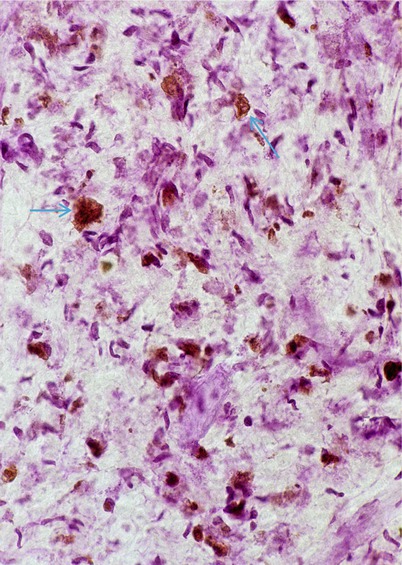
Fig. 20.6
Metachromatic leukodystrophy (MLD) inclusions (arrows) stained brown with acidified cresyl violet (Hirsh Pfeiffer stain) (400×)
The biochemical errors that cause MLD are dysfunction of arylsulfatase A, its activator protein (AB variant), or of multiple sulfatases. The ARSA gene has been fully characterized (Gieselmann et al. 1991). The fundamental difference between the various age-defined MLD subsets seems to be related to the amount of residual ARSA activity (Polten et al. 1991). In cell culture, fibroblasts from patients with adult-onset MLD are better able to clear sulfatide than those taken from infantile-onset patients (Percy et al. 1977). Polten and colleagues (1991) identified two important mutations of the ARSA gene: the “I” allele and the “A” allele. The former results in an unstable RNA that does not permit synthesis of a functional ASA, while the latter leads to synthesis of a rapidly degraded ASA protein (Polten et al. 1991). Patients with an I/I genotype have no functional ASA activity and present in infancy. Patients with the A/A genotype have sufficient enzyme activity to prevent presentation until early adult life, and patients with the I/A genotype are intermediate (Polten et al. 1991). The MLD pseudodeficiency gene is yet another ARSA allele which leaves sufficient enzyme activity to permit a normal phenotype, even if paired with an MLD allele (Penzien et al. 1993). Although combination of such alleles seem to give a satisfactory account for phenotypic variability, there are some pedigrees with phenotype variability which would be hard to explain on this basis (Clarke et al. 1989).
Pathogenetic mechanisms in MLD have been further examined in transgenic mice deficient in cerebroside sulfotransferase (CST). CST mice show impaired maintenance of Nav 1.6 sodium channels in mature nodes and migration of Kv1.2 channels from the paranodal to the juxtaparanodal region, resulting in the disappearance of Caspr and neurofascin 155 (NF155) clusters, essential components of the axo-glial junction (Eckhardt 2008; Dupree et al. 2005). In the peripheral nervous system, CST-deficient nerves extend axonal protrusions at the node of Ranvier, abnormal enlarged vesicles, and unusually short or absent Caspr and NF155 clusters (Hoshi et al. 2007). Additionally, the number of Schmidt–Lanterman incisures are increased and elevated levels of annexin II suggest structural changes at the Schmidt–Lanterman incisures (Eckhardt 2008; Hayashi et al. 2007).
A variety of treatment schemes have been developed for a number of lysosomal storage disorders (Miranda et al. 2013; Patil and Maegawa1 2013; Pierret et al. 2008). Enzyme replacement therapy (ERT) is undergoing clinical trials. Small molecules capable of crossing the blood–brain or blood–nerve barriers can be used as enzyme enhancers of diverse ASA mutants, either as pharmacological chaperones or as proteostasis regulators, to reduce the biosynthesis of sulfatides or target different affected pathways downstream of the primary ARSA deficiency. Hematopoietic stem cell transplantation using bone marrow-derived or umbilical cord blood-derived sources represents another therapeutic possibility, especially using cells which overexpress the ARSA gene. Bone marrow-derived mesenchymal stromal cells (BM-MSCs) have also been investigated as therapeutic agents (Miranda et al. 2013; reviewed in Patil and Maegawal 2013). BM-MSCs can provide the defective protein, secrete a number of neurotrophic factors and chemokines, have low immunogenicity, lack tumorigenic potential, and have a strong ability to home to injured tissues after being systemically injected. Some small molecule pharmacological chaperones may assist misfolded-prone mutant enzymes by assisting their ER folding and others may work on the lysosomal environment normalizing calcium levels (Patil and Maegawal 2013).
20.1.2 Globoid Cell Leukodystrophy (Krabbe Disease)
20.1.2.1 Clinical Manifestations
Globoid cell leukodystrophy (GLD, MIM#245200) is an autosomal recessively inherited disease caused by mutation in the galactosylceramidase gene (GALC) resulting in defective galactosylceramide-β-galactosidase activity causing accumulation of galactosylceramide in neural and nonneural tissues. Similar to MLD, an atypical form of GLD (MIM#611722) is produced by saposin A activator deficiency. Onset is usually in the first year of life with retarded development and irritability, rapid progression to spasticity, blindness, deafness, and decerebration, and death following within a few years of onset (Hagberg 1984). Some patients, usually those with onset later in childhood, have a more indolent course characterized by visual failure, ataxia, spasticity, and dementia and may survive into the second or third decade (Kolodny et al. 1991). Onset in adulthood is very rare (Hagberg 1984; Hedley-Whyte et al. 1988). Peripheral neuropathy is a usually a minor part of the infantile disease, but may be evidenced by diminished distal reflexes or hypotonia, and becomes more prominent with increasing age (Hogan et al. 1969; Kolodny et al. 1991; Dunn et al. 1969). Cases with neuropathy as the dominant manifestation have been described (Hedley-Whyte et al. 1988; Lyon et al. 1991). More often there is only electrophysiological evidence of neuropathy, marked conduction slowing being typical (Dunn et al. 1969; Suzuki and Grover 1970; Kolodny et al. 1991; Lyon et al. 1991). Diagnosis may be made by assay of β-galactosidase activity in cultured fibroblasts, leukocytes, or serum (Kolodny et al. 1991) or, more recently, from gene analysis.
20.1.2.2 Pathology
Globoid cell leukodystrophy derives its name in part from the prominent demyelination that afflicts the CNS and in part from the presence of “globoid” cells, multinucleated giant cells up to 120 μm in diameter containing PAS-positive material (Allen and de Veyra 1967; Suzuki and Grover 1970). Globoid cells are seen predominantly in the brain, often around vessels, and may be seen in nonneural tissues, but are not present in peripheral nerves. However, ultrastructural examination reveals that the material stored in globoid cells is the same as the storage product detected in the peripheral nervous system.
Light Microscopy
Light microscopy does not always give evidence of peripheral nerve disease. In a review of GLD, Lyon et al. (1991) noted that seven of ten biopsies showed segmental demyelination but that three were normal, and typical inclusions were said to be present in only three of the ten. However, it is unclear how closely the nerves were studied. Review of the case reports and small series described in the literature lead us to suspect that electron microscopy would reveal typical inclusions in most nerve biopsies (Bischoff and Ulrich 1969; Hedley-Whyte et al. 1988; Hogan et al. 1969; Lake 1968; Dunn et al. 1969; Martin et al. 1974; Lyon et al. 1991; Schlaepfer and Prensky 1972; Schochet et al. 1976; Sourander and Olsson 1968; Suzuki and Grover 1970; Thomas et al. 1984; Vital and Vallat 1987; Yunis and Lee 1972). Storage material was detected in nerve biopsy from a 23 week fetus, but not from a 20 week fetus (Martin et al. 1981).
The multinucleate PAS-positive cells seen in the CNS are not found in peripheral nerve. Some authors have described cells containing fine PAS-positive granules or sudanophilic material in the endoneurium, but this does not seem prominent (Suzuki and Grover 1970; Schochet et al. 1976; Schlaepfer and Prensky 1972; Allen and de Veyra 1967) and may not be distinguishable from control material (Bischoff and Ulrich 1969). No metachromatic material is detected, and Oil Red O staining is also negative (Dunn et al. 1969; Hogan et al. 1969). The substantial effort that went into characterizing the histochemical properties of the storage product is now of historical interest only (Sourander and Olsson 1968; Allen and de Veyra 1967).
Nerve biopsy in GLD reveals a demyelinating neuropathy, with some thinly myelinated fibers and a variable prominence of onion-bulb formations and ongoing demyelination. In some reports, active segmental demyelination with naked axons and prominent debris-filled macrophages has been emphasized (Dunn et al. 1969; Suzuki and Grover 1970), while in others the picture has been that of diffuse hypomyelination and an indolent pathology (Thomas et al. 1984; Vos et al. 1983). Axon numbers are mildly to moderately reduced in most cases, but Wallerian degeneration is typically not seen. Large myelinated fibers may at times be more severely depleted, and unmyelinated fibers seem to be spared (Martin et al. 1974; Schlaepfer and Prensky 1972). Debris-filled macrophages may be seen in the endoneurium, often aggregated around capillaries (Fig. 20.7). Under oil immersion, needlelike inclusions may be identified within these cells. Fiber teasing indicates that segmental demyelination with remyelination is the predominant pathological process.
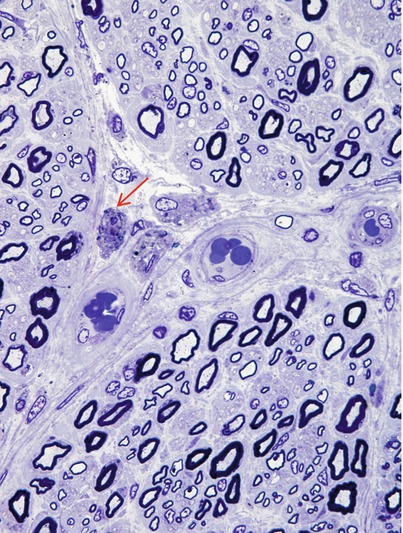
Fig. 20.7
Krabbe disease (globoid cell leukodystrophy, GLD). Perivascular macrophages (arrow) are accompanied by relatively large number of thinly myelinated axons (1 μm thick plastic section, 1,000×)
Electron Microscopy
Ultrastructural examination reveals the characteristic appearance of GLD inclusions: tubular or prismatic, straight or slightly curved, and elongated membrane-bound structures (Fig. 20.8a–c). Depending on the plane of section they range in appearance from polygonal or irregular optically empty clefts to hollow spicules as little as 10 nm in width. These optically empty clefts may contain a small amount of granular osmiophilic material or a few parallel lamellae of storage substance (Martin et al. 1974). Presumably most of the stored substance has been removed during tissue preparation. The inclusions seem to be rigid and very resistant to degradation, with needlelike clefts sometimes appearing to protrude into and even disrupt the plasmalemma of the cells containing them, becoming extruded into the extracellular space (Bischoff and Ulrich 1969). Inclusion material may be seen lying freely in the endoneurium (Thomas 1993). Typical GLD inclusions are seen in macrophages and in as many as half of myelinating and nonmyelinating Schwann cells (Bischoff and Ulrich 1969).
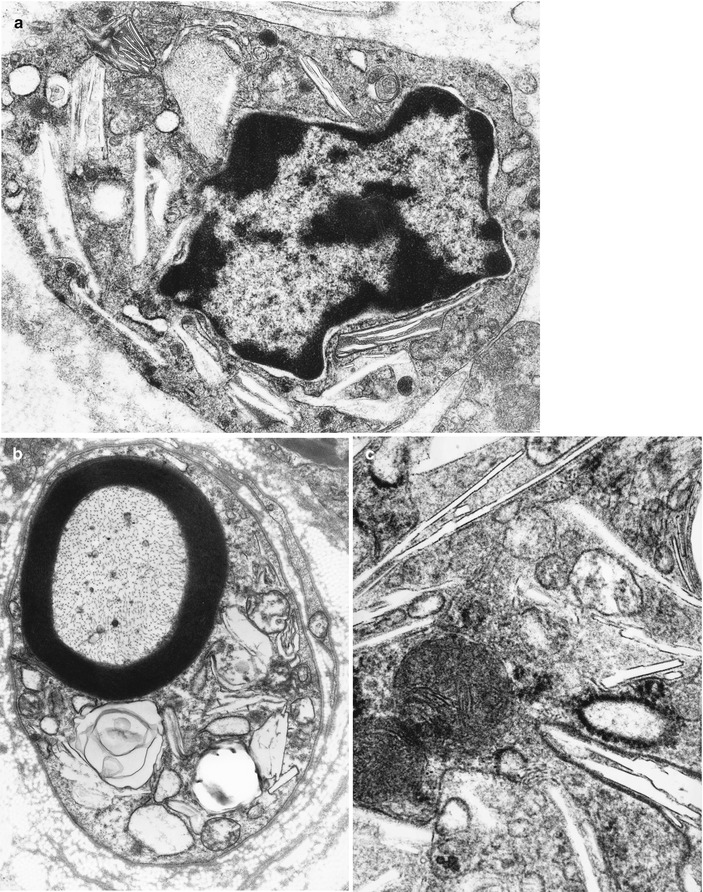
Fig. 20.8
Krabbe disease (GLD). Characteristic inclusions in macrophage (a) and Schwann cell (b) and at higher magnification in (c) (Magnification; a, 27,600×; b, 12,780×; c, 46,200×)
Some reports have described storage material within macrophages taking the form of cytosomes containing straight and twisted tubules, 30 nm in diameter (Schochet et al. 1976; Yunis and Lee 1972; Vos et al. 1983; Goebel et al. 1990). A similar substance has at times been found in CNS globoid cells and resembles the ultrastructural appearance of purified bovine cerebroside (Yunis and Lee 1972; Schochet et al. 1976).
20.1.2.3 Pathogenesis and Therapy
The mechanism of myelin damage and cell death in GLD remains uncertain (Suzuki and Suzuki 1989). Defective function of the enzyme galactosylceramide-β-galactosidase (also called galactosylceramidase) is always associated with GLD. Dogs and rats with the same defect have an identical disease (Suzuki and Suzuki 1989), and GLD affects only those tissues where galactosylceramide accumulates. Injection of cerebroside, but not other sphingolipids, into animal tissue produces typical globoid cells (Olsson et al. 1966). The “indigestibility” of the storage substance of GLD (as described above) may relate to the formation of multinucleate giant cells. A related sphingolipid, psychosine, also broken down by galactosylceramide-β-galactosidase, is consistently found to be increased in GLD tissue and is highly cytotoxic (Suzuki and Suzuki 1989). Various neuropathic mechanisms have been proposed in studies of the twitcher mouse model of GLD including activation of the pro-apoptotic protease caspase 3 in sciatic nerves (which occurs before demyelination), axonal instability, defective axonal transport, and deregulated ion channels including loss of Na+ channel concentration at the nodes of Ranvier (Siddiqi et al. 2006; Sakai 2009; Kagitani-Shimono et al. 2008; Smith et al. 2011). Psychosine rapidly accumulates in lipid rafts (White et al. 2009) and may interfere in multiple signaling pathways (Suzuki 1998). Thus, it is likely that the accumulation of galactosylceramide and its toxic metabolite psychosine causes the histologic features and tissue injury seen in GLD (Suzuki and Suzuki 1989).
As in other lysosomal disorders, experimental treatment methods have been investigated in human or animal models including hematopoietic stem cell transplantation, chaperone therapy, enzyme replacement therapy, deprivation of the substrate, gene therapy, and cytokine therapy (Sakai 2009). Siddiqi et al. (2006) found that nerve conduction studies improved in 7 (60 %) of 12 patients after hematopoietic stem cell transplantation followed for an average of 18 months.
20.1.3 Fabry Disease
20.1.3.1 Clinical Manifestations
Fabry disease (MIM#301500), also called angiokeratoma corporis diffusum in reference to the frequent presence of cutaneous angiokeratomas, is an X-linked disease resulting from a mutation in alpha–galactosidase A (GLA), has been localized to the long arm of the X chromosome, and codes for α-galactosidase A which in its full blown form affects only males, although heterozygote females may be mildly symptomatic (Morgan et al. 1990; Brady 1993; Biegstraaten et al. 2012). Age of symptom onset is usually childhood or adolescence, with diagnosis often delayed into adult life, the significance of skin lesions and corneal “whorl” dystrophy (Fig. 20.9) going unappreciated for many years (Morgan et al. 1990). Presentation with renal dysfunction or episodic painful paresthesia is typical, with cardiac and cerebral vascular disease representing important causes of morbidity and renal failure in mid-adult life being the usual cause of death prior to the availability of dialysis and renal transplantation. Autonomic features include hypo- or anhidrosis, reduced salivation and lacrimation, and gastrointestinal tract disordered motility; however, decreased sweating has been attributed both to autonomic neuropathy and to direct sweat gland involvement (Bersano et al. 2012). Routine clinical and electrophysiological testing for evidence of a peripheral neuropathy is often unrewarding, but specific assessment of small fiber function (Ohnishi and Dyck 1974; Morgan et al. 1990) and quantitative determination of numbers of axons within the epidermis (IENFD, Biegstraaten et al. 2012) will reveal impairment. Accumulation of glycolipid in DRG neuron cell bodies and their eventual loss may also play a role (Schiffmann 2006). Diagnosis is often suspected on the basis of the clinical picture and is confirmed by assay of the activity of the affected enzyme, α-galactosidase A (lysosomal hydrolase a-galactosidase A), in plasma or leukocytes. This is typically 10 % or less of the normal value in affected individuals and 50 % of normal value in heterozygote females. Molecular techniques may be used to demonstrate GLA, alpha–galactosidase A, gene defects.
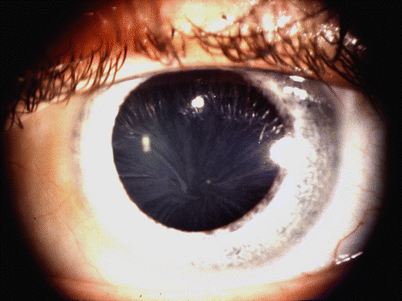
Fig. 20.9
Fabry disease cornea verticillata (“vortex keratopathy”) is characteristic of Fabry disease
20.1.3.2 Pathology
Nerve biopsy has minimal diagnostic value in Fabry disease because of the characteristic clinical manifestations and the presence of reliable noninvasive tests. If tissue is taken, the storage material of Fabry disease will be found predominantly in vascular cells: endothelial cells, smooth muscle cells, and pericytes; skin biopsy will readily demonstrate characteristic inclusions. However, when the diagnosis is unsuspected, nerve biopsy reveals pathognomonic histology, as has occurred once in our experience. Other detailed reports of nerve histology are available (Ohnishi and Dyck 1974; Kocen and Thomas 1970; Bischoff et al. 1968; Gemignami et al. 1984; Sima and Robertson 1978; Tabira et al. 1974; Fukuhara et al. 1975; Vital et al. 1984; Tome et al. 1977; Pellissier et al. 1981; Schiffmann 2006).
Light Microscopy
Light microscopy invariably reveals an accumulation of storage material in various cell types. The inclusions are birefringent, and a “Maltese cross” pattern can be readily demonstrated on polarized light examination of fresh frozen sections (Fig. 20.10). Frozen sections, and less reliably paraffin-embedded material, show Sudan Black B, Oil Red O, LFB, and PAS positivity, with the latter reduced by digestion with diastase (Fukuhara et al. 1975; Kocen and Thomas 1970; Hashimoto et al. 1965). The storage material is partially dissolved in the process of paraffin embedding, and H&E staining of processed material often shows little pathology (Fig. 20.11a), but exposure of formalin-fixed tissue to 3 % potassium chromate for 1 week reduces this problem (Desnick and Bishop 1989). Osmicated toluidine blue-stained plastic-embedded sections showing relatively well-preserved axonal population (Fig. 20.11b) circumvent this problem, and the inclusions, typically 1–2 μm in diameter, can be readily seen in endothelial cells, pericytes, smooth muscle cells, perineurial cells, and fibroblasts, but usually not in Schwann cells or axons (Fig. 20.11c–e). Endothelial cells seem to be particularly distended with the storage material.
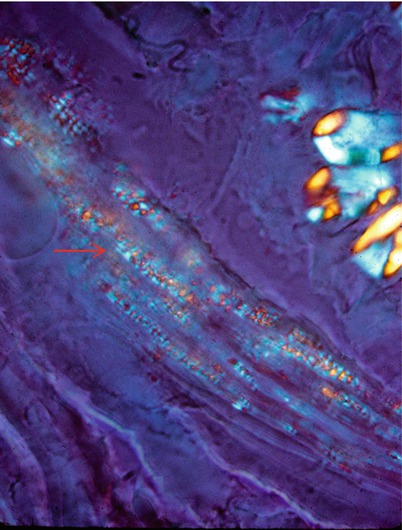
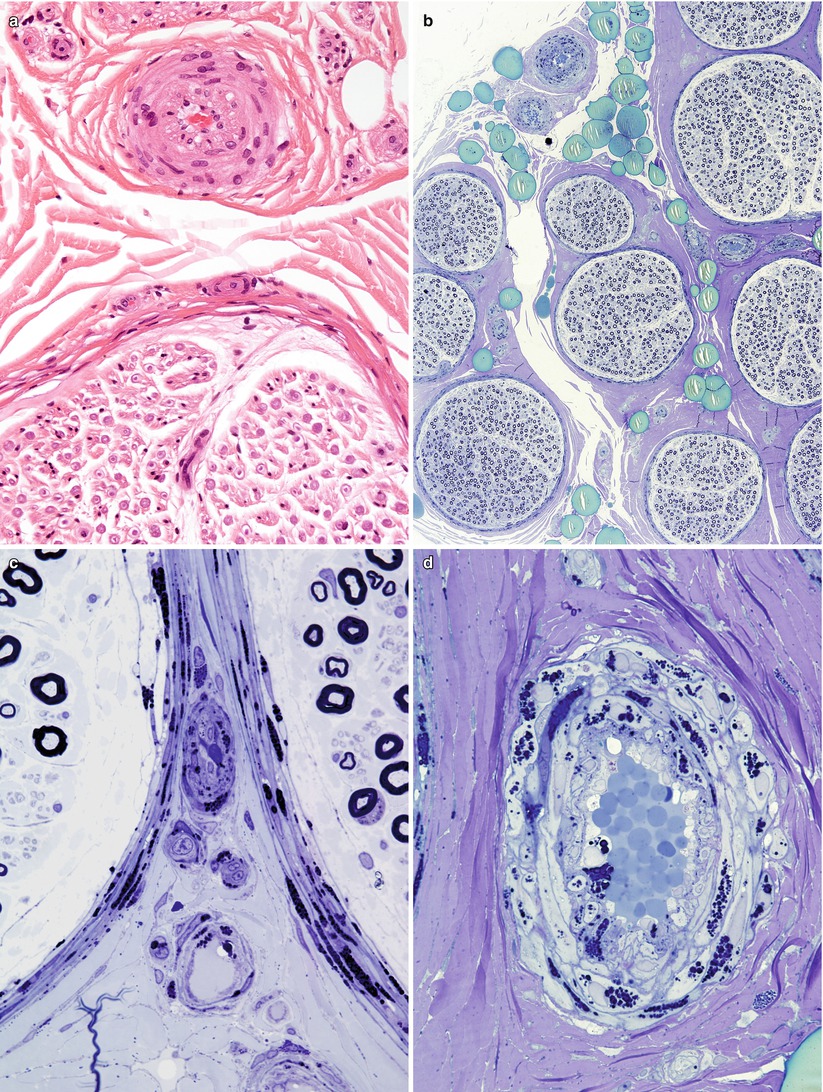
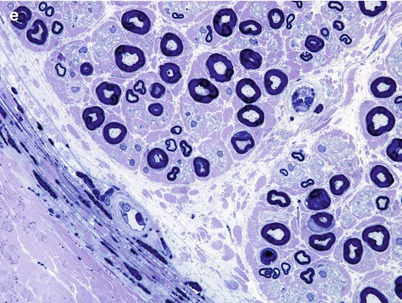
Fig. 20.10
Fabry disease: polarization microscopy discloses “Maltese cross” birefringence in perineurium (arrows). Unstained fresh frozen tissue (400×)
Fig. 20.11
Fabry disease: (a) H&E-stained section of small epineurial artery shows only subtle involvement. (b) All fascicles are equally affected and show relatively small loss of axons. (c–e) Note the large quantity of osmiophilic lipid within the cytoplasm of perineurial, endothelial, and endoneurial macrophages (1 μm thick plastic sections; magnifications: a, 400×; b, 10×; c–e, 1,000×)
The classic peripheral nerve alteration in Fabry disease is said to be a relatively selective loss of small myelinated and perhaps unmyelinated fibers (Ohnishi and Dyck 1974; Kocen and Thomas 1970; Gemignami et al. 1984). However, this was not seen in our material (Case 20.1) (Figs. 20.11 and 20.12), and a number of literature reports describe large myelinated or pan-myelinated fiber depletion (Fukuhara et al. 1975; Pellissier et al. 1981; Vital et al. 1984; Tabira et al. 1974). Active degeneration is rarely seen due to the indolent nature of the process, and regenerating clusters can even be prominent (Gemignami et al. 1984). It is possible that concomitant renal failure accounts for depletion of large myelinated fibers, but a predominantly large fiber neuropathy has been seen in Fabry patients with minimally abnormal or normal renal function (Fukuhara et al. 1975; Pellissier et al. 1981, Case 20.1). Demyelination is not a significant element of the neuropathy of Fabry disease, although teased fibers may show some demyelinated–remyelinated internodes, presumably of a secondary nature (Gemignami et al. 1984; Ohnishi and Dyck 1974).
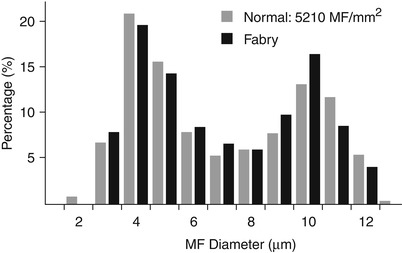
Fig. 20.12
Fabry disease: slight decrease in fiber density for age without loss of small myelinated axons
Electron Microscopy
Ultrastructural examination reveals the characteristic appearance of the osmiophilic inclusions. Although acid phosphatase staining indicates that the storage material is contained within lysosomes (Hashimoto et al. 1965), a limiting membrane is sometimes not seen. The electron-dense inclusions may be rounded (particularly in perineurial cells, Fig. 20.13a, b), have a comma-like shape (Fig. 20.14), or take the form of “zebra” bodies. At high magnification a 4–7 nm periodicity is commonly seen, and other patterns include regular arrays of concentric rings or hexagons, or amorphous electron-dense material with no visible periodicity (Fig. 20.14). Most studies have not described inclusions in Schwann cells or axons, but a few authors have illustrated examples of inclusions in these cells (Sima and Robertson 1978; Perrelet et al. 1969; Vital and Vallat 1987, Fig. 251).
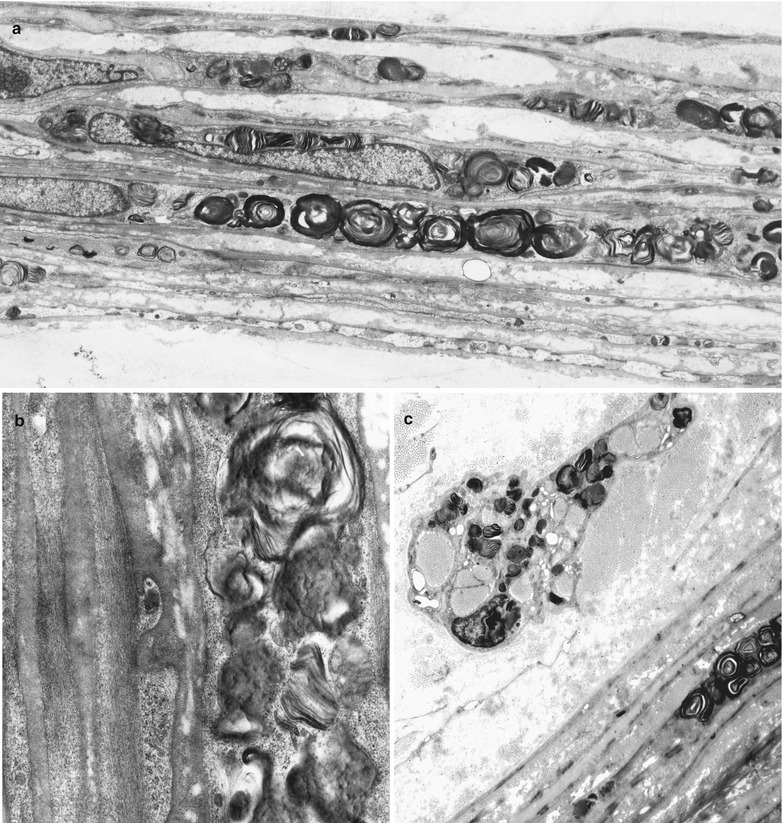
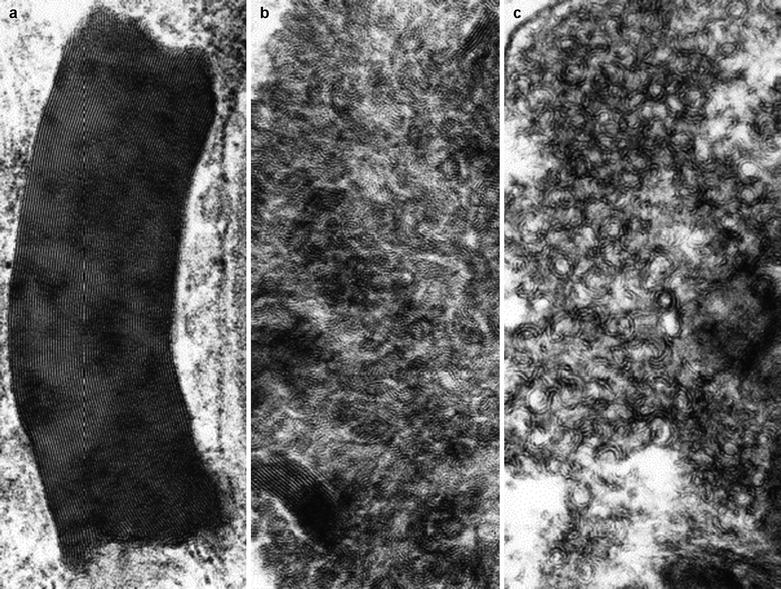
Fig. 20.13
Fabry disease: lipid storage in perineurium (a, b) and endoneurial macrophage (c) (a, 3,840×; b, 19,760×; c, 6,000×)
Fig. 20.14
Fabry disease: the variable morphology of stored lipid is illustrated (Magnification: a, 165,600×; b, c, 164,800×)
Deposition of inclusion material in endothelial cells can be very impressive, but rarely occurs to such an extent that the lumen is occluded (Fig. 20.15a, b). Unmyelinated fibers may show active degenerative changes more frequently than in controls, and the number of denervated nonmyelinating Schwann cell subunits may be increased (Sima and Robertson 1978; Ohnishi and Dyck 1974). A histogram of unmyelinated fiber counts demonstrates a shift in size towards small UF diameters (Pellissier et al. 1981).
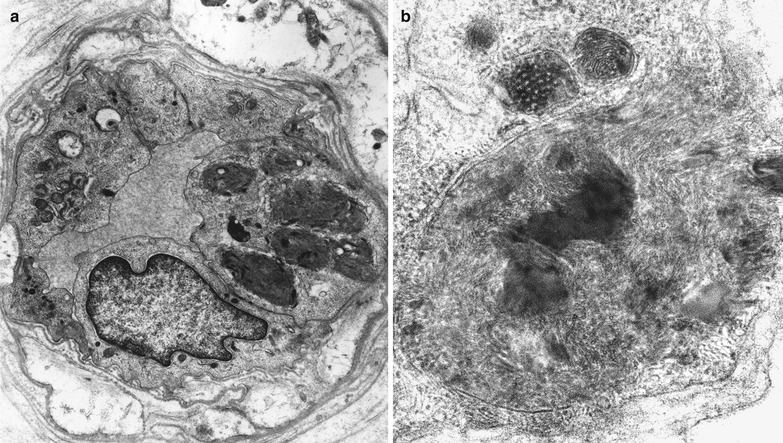
Fig. 20.15
Fabry disease: epineurial vessels showing lipid accumulation in endothelial and smooth muscle cells (Magnification: a, 6,250×; b, 20,000×)
20.1.3.3 Pathogenesis and Therapy
The defective gene (GLA, alpha–galactosidase A, MIM#300644) has been localized to the long arm of the X chromosome and codes for α-galactosidase A. A number of different deletions and point mutations causing an abnormally formed or an abnormally processed protein have been found in patients with Fabry disease (Bernstein et al. 1989; Lemansky et al. 1987). Precisely how the genetic defect relates to disease manifestations is unclear. The peripheral nerve disease is thought to be a neuronopathy preferentially involving small myelinated and unmyelinated axon cell bodies (Ohnishi and Dyck 1974; Gemignami et al. 1984; Morgan et al. 1990). Alternatively, the prominent vascular involvement seen in Fabry disease has been suggested as a cause of peripheral nerve dysfunction (Fukuhara et al. 1975).
Enzyme replacement therapy and small molecule chaperones have increased residual enzyme activity in animal models and cultured cells from Fabry disease patients (Bersano et al. 2012; Toyooka 2011, 2013). Nonetheless, the IENFD was unchanged after 12–18 months of enzyme replacement treatment (Schiffmann et al. 2006), although there was evidence of improved small nerve fiber function (Hilz et al. 2004).
Case 20.1
A 34-year-old male of Northern Irish descent was referred because of headache and transient visual obscurations. One year previously he had experienced transient weakness of the left arm and leg lasting several days. Additional questioning revealed a history dating to the teen years of undiagnosed sharp shooting pains in the legs and arms, and a childhood appendectomy for abdominal pain which revealed a normal appendix. The patient’s family included four brothers (one nonidentical twin) and one sister, all of whom were reportedly well. The parents were also apparently unaffected. Neurological examination was essentially normal, but ophthalmological opinion was requested because of suspected mild papilledema of the left optic disk. The consultant ophthalmologist did not feel that papilledema was present, but noted corneal opacification and raised the possibility of Fabry disease at that time. Fluorescein angiography was normal.
The patient was admitted to hospital 5 months later with sudden onset of near-total vision loss in the left eye. A diagnosis of optic neuropathy was made, but the question of an inflammatory or vascular basis remained unclear; the complete lack of improvement favored the latter. Note was made of “whorl keratopathy” of the cornea. Erythematous papules were observed on the backs of the shoulders and in the flanks and groin area. Buccal and labial telangiectasias were also noted. A neurological examination at this time revealed mild right arm and leg weakness, diffuse hyperreflexia, and bilaterally upgoing toes. His course over the next 2 years was notable for urinary difficulties ultimately requiring intermittent catheterization. Although the diagnosis of Fabry disease was established biochemically by this time, nerve biopsy was prompted by suspicion of a diagnosis of polyglucosan body disease.
Laboratory testing included normal renal function and normal 2-D echocardiogram. CSF examination revealed a slight pleocytosis on two occasions 6 months apart (9, 17 cells, respectively, predominantly lymphocytes), and normal protein, glucose, and opening pressure. Oligoclonal banding was negative. CT and MRI of the head were normal on three separate occasions. Serum α-galactosidase was 22.5 ng/mg/h (normal >100), with increased urinary ceramide trihexoside excretion. Nerve conduction and EMG studies were normal. Small fiber function and sweating were not specifically tested.
20.1.4 Niemann–Pick Disease
20.1.4.1 Clinical Manifestations
The Niemann–Pick (NP) group of diseases is characterized by tissue storage of sphingomyelin and/or cholesterol (Spence and Callahan 1989). All subtypes are inherited as autosomal recessive traits. Niemann–Pick disease is currently subdivided on the basis of biochemical and molecular criteria into several separate classes: types A (MIM#257200) and B (MIM#607616) (both type I in older classification) are deficient in acid sphingomyelinase and are sphingomyelin storage diseases (Takahashi et al. 1992), and types C and D (MIM#257220) (both previously type II) are deficient in the NPC1 protein and represent lysosomal disease of intracellular cholesterol trafficking. Foam cell infiltration and visceromegaly are common to all, but severe neurological involvement occurs only in types A (a rapidly progressive neurodegenerative course leading to death by 2–3 years of age) and C and not in type B (McGovern and Desnick 2011).
Hepatosplenomegaly is a central element of the NP clinical picture, and biopsy reveals “foam cells,” macrophages distended with storage material ultrastructurally similar to that described below. Peripheral neuropathy is usually not a clinically significant part of the disease (Gumbinas et al. 1975). Nearly all the NP patients with neuropathy discussed below had conduction slowing suggestive of a demyelinating neuropathy.
Diagnosis traditionally has been made through clinical suspicion and the histologic demonstration of “foam cells” on bone marrow or liver biopsy. More recently assays for sphingomyelinase in tissue culture or fluids have been replaced by genetic studies. Niemann–Pick type A is caused by mutation in the sphingomyelin phosphodiesterase-1 gene (SMPD1), which encodes acid sphingomyelinase (ASM; Schuchman et al. 1992) and Niemann–Pick disease types C and D are caused by mutation in the NPC1 gene. Recently a number of small molecule agents including cyclodextrin have been used to improve visceral and neurodegeneration (Perez-Poyato and Pineda 2011; Liu et al. 2010).
20.1.4.2 Pathology
Clinicopathological studies of peripheral nerve in NP are rare (Landrieu and Said 1984; Da Silva et al. 1975; Gumbinas et al. 1975; Hahn et al. 1994; Elleder 1989; Elleder et al. 1986; Babel et al. 1970 p 130; Bischoff 1979; Dubois et al. 1990). Most reports have considered NP type A except those of Elleder (1989) and Hahn et al. (1994). Our experience consists of three nerve biopsies in a brother and sister with NP type A who survived well into adult life (Case 20.2). Two biopsies in the sister were taken 15 years apart.
Light Microscopy
Sural nerves from patients with Niemann–Pick type A show generalized attenuation of myelin sheaths (Fig. 20.16a, b). Classic “foam cells” are sometimes observed in the endoneurium (Gumbinas et al. 1975). These large cells are often found in a perivascular distribution and demonstrate a finely multivacuolated cytoplasm (Fig. 20.16c). Light microscopy provides sufficient resolution to detect some Schwann cell inclusions, about 1 μm in size. With cryostat sections, the storage material is said to display “Maltese cross” birefringence similar to that of Fabry disease (Elleder 1989), although we have not detected this in our material. Oil Red O-positive material is seen on both frozen and paraffin-embedded sections, and the storage substance is particularly well highlighted with Giemsa staining. Some of the lipid inclusions are autofluorescent.
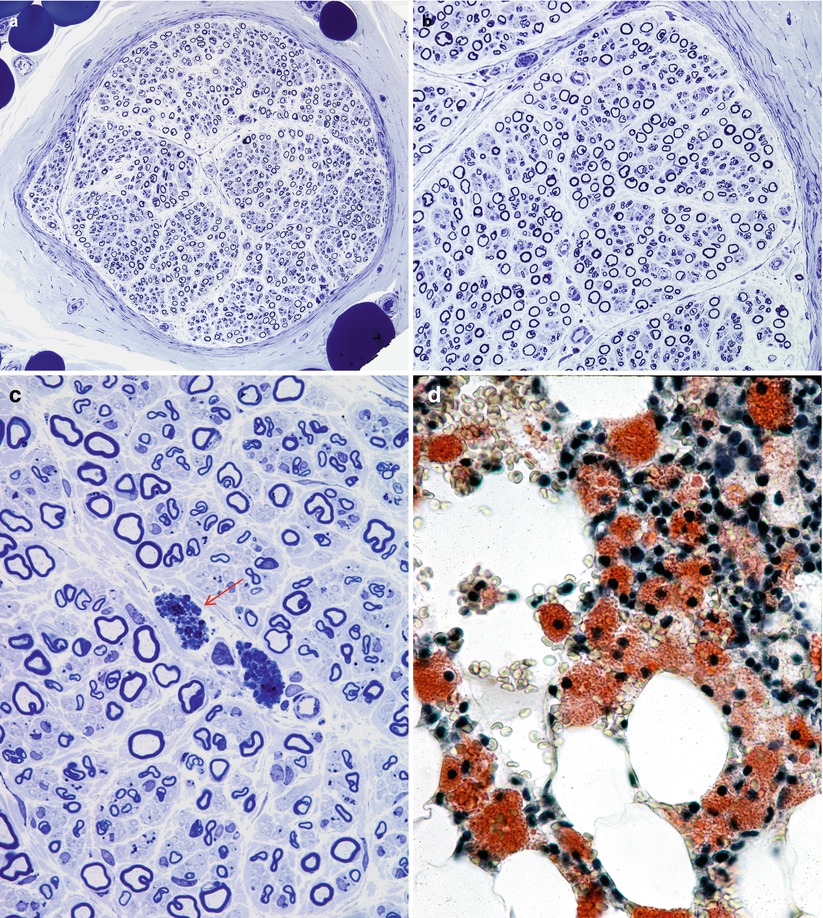
Fig. 20.16
Niemann–Pick disease, type A: note universal attenuation of myelin sheaths (a, b), lipidized perivascular endoneurial macrophages (arrow, c) and foamy bone marrow histiocytes (d). (a–c, 1 μm thick plastic section; d, frozen section, Oil Red O stain) (Magnifications: a, 400×; b, 600×; c, 1,000×; d, 1,000×)
Axon numbers are minimally or not at all reduced in most literature reports, and in our experience, even with the passage of many years (Dubois et al. 1990; Hahn et al. 1994; Landrieu and Said 1984). In two nerve biopsies from the same patient taken at the ages of 35 and 50, there was no evidence of any histological progression of the disease (Fig. 20.17a–c). A picture of excessively thin myelin sheaths is most common (Fig. 20.16a–c), with naked axons and active demyelination being quite rare (Gumbinas et al. 1975; Hahn et al. 1994; Landrieu and Said 1984; Da Silva et al. 1975). Onion-bulb formations were present in our material, but have not been reported otherwise. The two patients reported by Dubois et al. (1990) had no light microscopic evidence of neuropathy, but ultrastructural examination revealed characteristic storage material in Schwann cells.
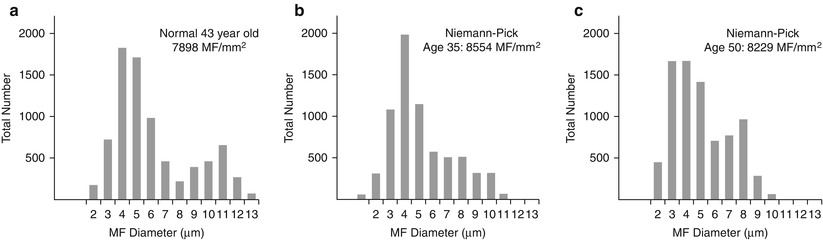
Fig. 20.17
Two biopsies spanning 15 years show a normal fiber density (b, c) as compared to an age-matched control (a)
Electron Microscopy
The typical ultrastructural finding in NP type A is an accumulation of membrane-bound inclusions, most prominently in Schwann cells of myelinated and unmyelinated fibers (Fig. 20.18) and in histiocytes (Fig. 20.19a, b). In reported cases the storage material has taken a variety of appearances, including concentric whorls of loosely packed membranous material, amorphous or granular electron-dense material in which a subtle periodicity may be seen, single or multiple lucent vacuoles, or “zebra” body-like inclusions. Some of the stored material is indistinguishable from lipofuscin and ceroid (Figs. 20.18 and 20.19a, b), and as Elleder (1989) points out, a major part of the storage material found in the reticuloendothelial system is indeed lipopigment, especially in the late-onset disease variants (Spence and Callahan 1989). In our patients the stored substance was most often amorphous and lipofuscin-like, and “zebra bodies” were not seen. However, at times osmiophilic regions showing a periodicity of 4–6 nm emerged from the amorphous background (Fig. 20.19b). The foamy cells seen on LM are revealed as macrophages containing storage material similar to that seen in the Schwann cells (Fig. 20.19a). Inclusions can also be seen in endothelial cells, pericytes, myocytes, perineurial cells, bone marrow, and fibroblasts (Fig. 20.20a, b). Axonal changes are less obvious and nonspecific and include loss of filaments, mitochondrial accumulations, increases in axoplasmic vesicles, and numerous dense bodies (Babel et al. 1970; Landrieu and Said 1984). Babel et al. (1970, Fig. 62) illustrate a membrane-bound axonal inclusion composed of lamellated material with a periodicity of 5.6–6 nm, but lamellated intra-axonal cytosomes are a nonspecific finding and might not relate to the storage process occurring in Schwann cells.
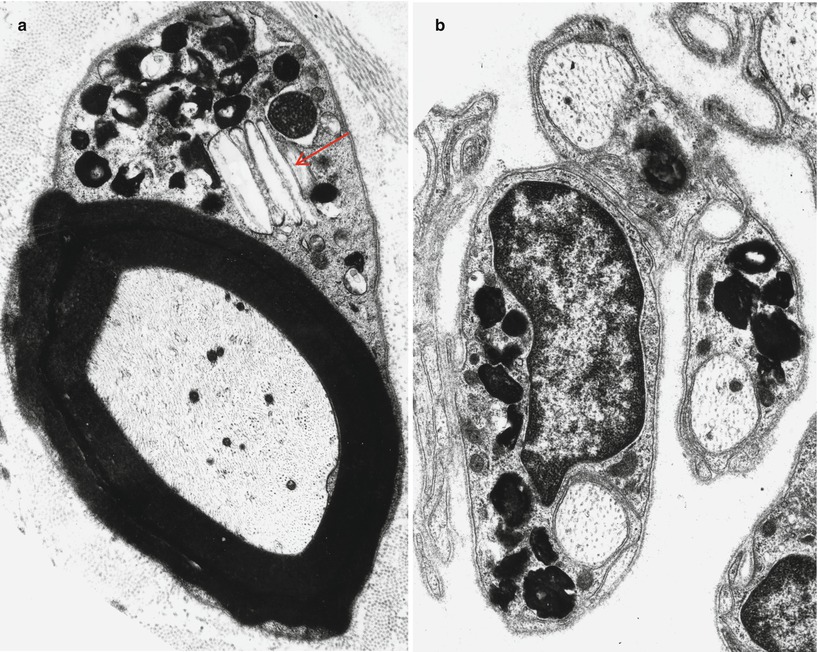
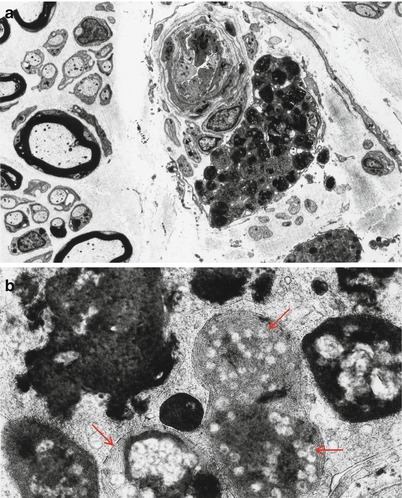
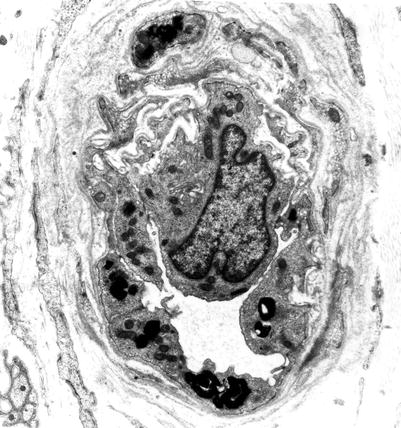
Fig. 20.18
Niemann–Pick type A: lipid inclusions, most of which are indistinguishable from lipofuscin, are seen in the cytoplasm of myelinating and nonmyelinating Schwann cells. Note Pi-body (arrow, a) (Magnification: a, 11,172×; b, 16,188×)
Fig. 20.19
Niemann–Pick type A: perivascular endoneurial macrophage laden with lipid debris. In (b) regions of concentric periodicity (arrows) are intermingled with amorphous material of variable osmiophilia (a, 3,040×; b, 45,144×)
Fig. 20.20
Niemann–Pick type A: lipid inclusions in endothelial cells and pericytes (Magnification: 7,040×)
In NP type C, axonal alterations seem to be a central part of the histological picture (Hahn et al. 1994; Elleder 1989), but the paucity of available material precludes generalization. An accumulation of whorled and multi-lobulated flocculent material in most endoneurial cellular components is described by Hahn et al. (1994). However, these authors emphasize the presence of axonal alterations, with occasional “… accumulation and condensation of neurofilaments, vesicular profiles, dense bodies, and abnormal mitochondria resembling changes of neuroaxonal dystrophy” (Hahn et al. 1994). Elleder (1989) has also commented on “… neuroaxonal dystrophy of a nonspecific type …” in cutaneous nerve axons. Skin biopsy in NP type C reveals lysosomes containing dense material or loosely packed lamellated material in a clear background in axons and Schwann cells, as well as fibroblasts, epithelial cells, and pericytes (Boustany et al. 1990). Curvilinear inclusions in endothelial cells described by Hahn et al. (1994) appear similar to those seen in neuronal ceroid lipofuscinosis.
Case 20.2
A 37-year-old man was referred with a history of dementia and ataxia. At the age of 3 he was found to have an enlarged spleen. A psychiatric disorder with paranoid features emerged during his teen years, and in his 20s, he became frankly demented. Progressive gait unsteadiness followed by limb ataxia also appeared in the second decade. Examination at the age of 37 revealed severe dementia, no significant weakness, preservation of reflexes, grossly intact sensation, and ataxia of all limbs. Nerve conduction studies showed normal amplitudes, with slightly prolonged distal latencies and conduction velocity of 30 m/s in both motor and sensory nerves, suggesting a mild demyelinating neuropathy. CT scan showed severe cerebellar atrophy affecting the vermis more than the hemispheres.
The patient’s sister presented with gait difficulty in her teens, progressing very slowly to walker dependence by the age of 40. She was also thought to be cognitively impaired and demonstrated impulse control difficulties requiring treatment with neuroleptics. Examination of at the age of 50 years revealed mild mental impairment, normal fundoscopy, normal extraocular movements, and a slight dysarthria. The limbs displayed normal tone and strength throughout, with all reflexes reduced but obtainable, and no significant sensory loss to pin or vibration. Mild ataxia was present on heel–shin testing, but the gait was wide based and grossly ataxic. Hepatosplenomegaly was noted. Nerve conduction studies performed at the age of 35 showed a very mild neuropathy with amplitudes and conduction velocities just below the lower limit of normal.
Liver biopsy was performed on the brother and demonstrated numerous foamy histiocytes. Lipid analysis of the liver biopsy material revealed an accumulation of sphingomyelin. Cultured fibroblast sphingomyelinase activity in the sister was 1.1 nmol/mg/h (10 % interdecile range 8.6–289 nmol/mg/h). The brother’s cultured fibroblast and serum sphingomyelinase activities were also less than 10 % of the lower limit of normal. Nerve biopsies were performed on the brother at the age of 38, and on the sister at the ages of 35 and 50 (Figs. 20.16, 20.17, 20.18, 20.19, and 20.20).
20.1.5 Farber Disease (FD, Lipogranulomatosis)
20.1.5.1 Clinical Manifestations
Farber disease is a rare autosomal recessive infantile disease (MIM#228000) caused by mutation in the ASAH1 gene encoding acid ceramidase resulting in the deficiency of acid ceramidase, necessary for metabolism of the sphingolipid ceramide (Moser et al. 1989; Zappatini-Tommasi et al. 1992; Alayoubi et al. 2013). The characteristic clinical features are disseminated nodular subcutaneous, periarticular swellings and vocal cord thickening. The central nervous system is affected in about half of the patients. Peripheral nerve involvement is common, manifesting with hypotonia and atrophy, and nerve conduction studies have shown significant slowing (Fujiwaki et al. 1992; Pellissier et al. 1986). The usual outcome is rapid progression to death in the first few years of life, although some patients survive into the third decade (Moser et al. 1989).
20.1.5.2 Pathology
Histologic examination of the nodules of Farber disease reveals a granulomatous infiltrate, sometimes with mature granuloma formation. Storage material may take on a variety of ultrastructural appearances including Farber bodies, “banana” bodies, and “zebra” bodies (Zappatini-Tommasi et al. 1992; Burck et al. 1985). Farber bodies are curved tubular structures 14–17 nm in diameter found in great numbers within membrane-bound vacuoles or free within the cytoplasm and believed to correspond to stored ceramide (Rauch and Aubock 1983; Takahashi and Naito 1985; Pellissier et al. 1986). They bear a slight resemblance to the curvilinear profiles of Batten–Kufs disease, but are smaller and tubular, not lamellated. Farber bodies are seen in fibroblasts, histiocytes, and endothelial cells in extraneural tissues. Zebra bodies have been described in endothelial cells (Rauch and Aubock 1983).
A few nerve biopsy specimens have been closely studied (Vital and Vallat 1987; Vital et al. 1976; Pellissier et al. 1986; Rauch and Aubock 1983; Burck et al. 1985). Light microscopic examination of nerves has shown widespread hypomyelination and may reveal mild loss of large axons not sufficient to reduce the total MF density below normal (Fig. 20.21a) (Pellissier et al. 1986). Vital et al. (1976) indicated that unmyelinated fibers were spared in their specimen, but this was not quantitated. High-power LM can reveal pathognomonic vacuoles in myelinating Schwann cells (Fig. 20.21b). Frozen sections did not reveal any sudanophilic or metachromatic material in the case of Vital et al. (1976).
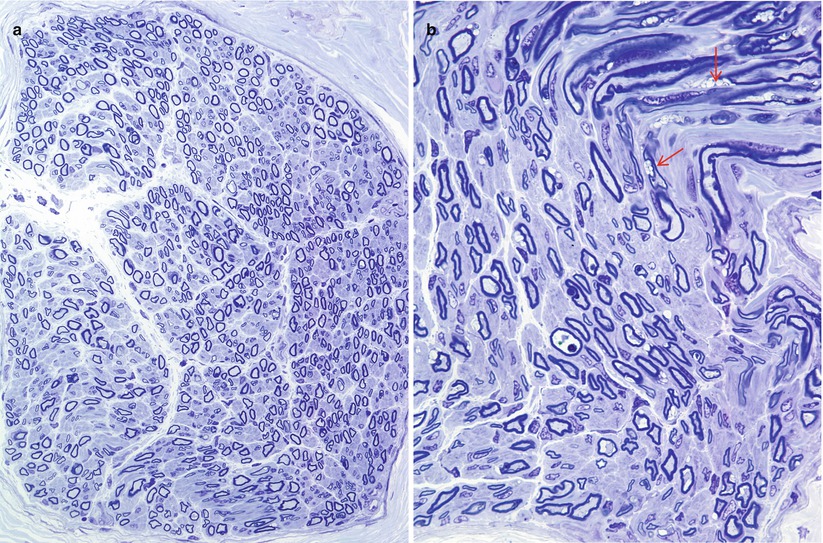
Fig. 20.21
Farber disease: note normal MF density (a), attenuated myelin sheaths, and accumulation of optically empty inclusions in Schwann cell cytoplasm (arrows, b) (1 μm thick plastic sections; magnification a, 400×; b, 1,000×) (Tissue courtesy of Dr. V. Jay)
Ultrastructural examination is diagnostic (Fig. 20.22). Myelinating Schwann cells contain electron-lucent membrane-bound clefts, which take a variety of shapes ranging from needlelike to elongated rectangular, angulated, polygonal, and even annular profiles (Fig. 20.23a, b). These may be up to 5 μm in diameter, may indent the axon, and are almost exclusively seen in myelinating Schwann cells (Rauch and Aubock 1983; Schmoeckel and Hohlfed 1979). Presumably the content of these structures is lipid, washed out in preparation. At times there is a residual content which Pellissier et al. (1986) described as “… either homogenous and osmiophilic, or less dark with an electron-dense lamellar component.” We have also observed, in nonmyelinating Schwann cells only, novel membrane-bound elongated bodies containing a central longitudinal striated region surrounded by amorphous electron-dense material (Fig. 20.24).
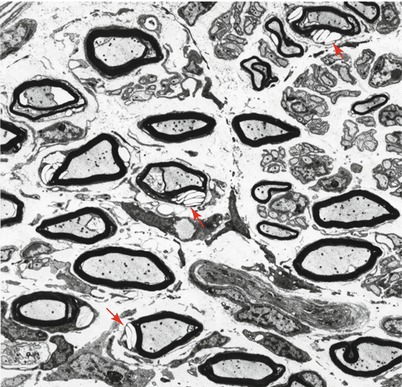
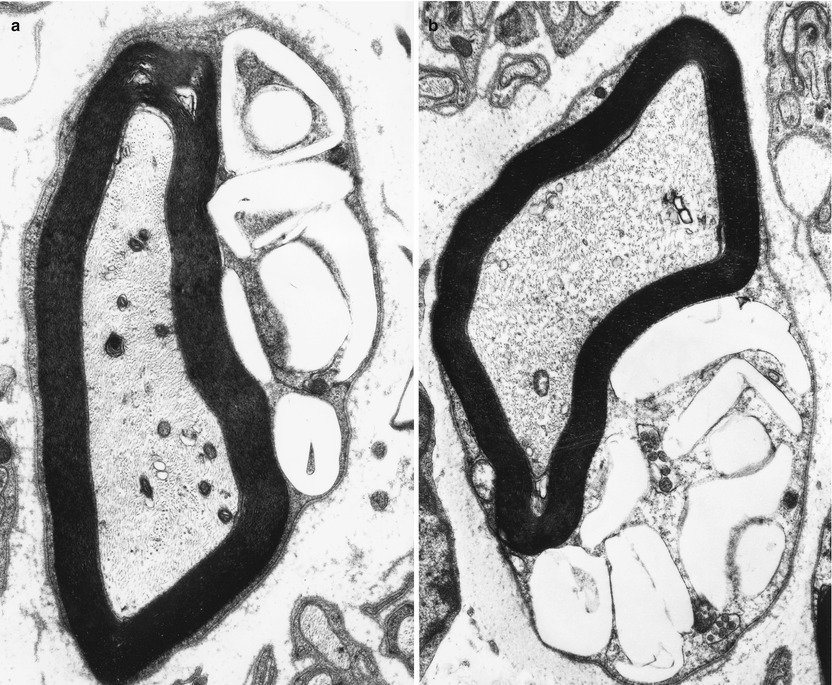
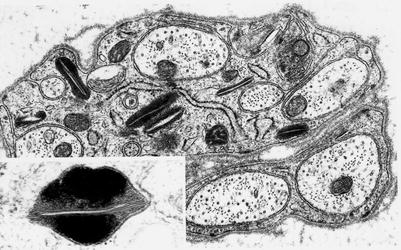
Fig. 20.22
Farber disease: numerous “banana” bodies are shown in myelinating Schwann cells (arrows) (4,000×)
Fig. 20.23
Farber disease: typical morphologies of “banana” bodies (a, 14,910×; b, 12,320×)
Fig. 20.24
Farber disease: nonmyelinating Schwann cells contain membrane-bound elongated striated bodies (20,080×, inset 91,200×)
20.1.5.3 Pathogenesis and Therapy
A mouse model has been developed (Alayoubi et al. 2013) via “knock-in” of a known human ASAH1 mutation into a conserved murine Asah1 gene locus. In that model it has been determined that the accumulation of ceramide prompts the release of MCP-1 which recruits circulating monocytes to tissues where they accumulate and fail to degrade the sphingolipids and ceramide they engulf, amplifying the foamy appearance which is characteristic of FD. Neonatal gene therapy using recombinant lentivectors encoding the human acid ceramidase gene has shown a salutary effect in this experimental model. In patients allogeneic bone marrow transplantation has shown success in reducing the peripheral symptoms in Farber disease but failed to improve neurological symptoms and patient lifespan (Ehlert et al. 2007).
20.1.6 Other Sphingolipidoses
Examination of nerve biopsy has been carried out in a number of other sphingolipidoses, but the available peripheral nerve material is too scanty for detailed discussion. For completeness, these are discussed briefly.
GM2 gangliosidosis (MIM#272800) is caused by defects in hexosaminidase activity (Table 20.1) and manifests with a wide spectrum of infantile- and adult-onset diseases, the most common of which is Tay–Sachs disease (Federico et al. 1991a). Peripheral neuropathy is not a significant part of the disease, but motor neuronopathy is common in late-onset cases. The characteristic storage inclusions, seen best in neuronal perikarya, are membranous cytoplasmic bodies. Sural nerve biopsy in some of the adult-onset cases has shown a reduction in myelinated fiber density and evidence of axonal regenerative activity (Mitsuo et al. 1990; Mitsumoto et al. 1985; Federico et al. 1986, 1991a). Unfortunately, these case reports do not mention ultrastructural findings, and there are no well-documented reports of inclusions in sural or other peripheral sensory nerve biopsies. Severe destruction of myelin sheaths and axonal swellings were seen in a light microscopic study of sciatic nerve in Tay–Sachs disease, but no EM examination was performed (Kristensson et al. 1967). In the infantile (Tay–Sachs) and juvenile (Sandhoff) variants of GM2 gangliosidosis, intra-axonal membranous cytoplasmic bodies, “zebra” bodies, and dense amorphous inclusions have been observed in terminal myelinated and unmyelinated nerve fibers present in skin and muscle specimens (Schmitt et al. 1979; Abe et al. 1985; Dolman et al. 1977; Thomas et al. 1989; Burck et al. 1979). Bischoff (Fig. 4.61) illustrates unmyelinated fibers containing osmiophilic multilamellated inclusions in a patients with Tay–Sachs disease, but the tissue source is not specified (Bischoff 1979). Recent work with pharmacologic chaperones results in the protection of the mutated protein from degradation, increasing its lysosomal quantity and metabolic function (Chiricozzi et al. 2013). Chaperone therapy has been attempted in GM1 gangliosidosis, Fabry disease, Gaucher disease, and Morquio B disease (Chiricozzi et al. 2013).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


