and Robert E. Schmidt2
(1)
Sunnybrook and St Michael’s Hospitals, University of Toronto, Toronto, ON, Canada
(2)
Division of Neuropathology Department of Pathology, Washington University School of Medicine, St. Louis, MO, USA
Peripheral neuropathy caused by a genetic defect is common in clinical practice as the manifestation of a variety of inherited disorders. The polyneuropathy may occur as the sole expression of the genetic defect or may be seen in association with the involvement of the CNS or as part of a complex multisystem disorder. Many of these diseases have a relatively benign and slowly progressive course, with most patients leading a nearly normal life. Recent advances in molecular genetics have reduced the utility of nerve biopsy in the diagnosis of genetically determined neuropathies and allow for the beginnings of a rational classification. The full gamut of genes responsible for inherited neuropathies is now so extensive that it has spawned its own database “The Inherited Peripheral Neuropathies Mutation Database” (http://www.molgen.ua.ac.be/CMTMutations/). We will not attempt to catalogue the pathology of each of these mutations; rather, we will illustrate distinctive neuropathy phenotypes which characterize multiple different genotypes. For additional information the reader is directed to a number of excellent recent reviews (Reilly et al. 2011; Klein et al. 2013; Rossor et al. 2013; Saporta and Shy 2013; Sagnelli et al. 2013). Inherited neuropathies with accumulation of storage material are discussed in Chap. 20. With the exception of clear-cut familial occurrence, often not demonstrable, the single most important clue to the presence of a genetically determined neuropathy is duration of illness. While some neuropathies can manifest episodically or acutely (e.g., porphyria), the vast majority have an insidious onset and progress over many years, except for hereditary neuralgic amyotrophy.
A heterogeneous group of neuropathies is classified under the rubric of Charcot–Marie–Tooth (CMT), previously known as Hereditary Motor and Sensory Neuropathy (HMSN), and in practice these account for the majority of genetically determined neuropathies. Peripheral nerve dysfunction associated with cerebellar or spinocerebellar disease is next in frequency.
19.1 Charcot–Marie–Tooth and Cognate Disorders
The prevalence of CMT has been estimated at 5–41 per 100,000 (Dyck et al. 1993; Berciano and Combarros 1990). This wide range is largely due to the fact that many cases are mild and patients never require medical attention. Dyck has estimated that only 10 % of individuals with CMT-1 seek a physician for symptoms relating to the disease (Dyck et al. 1993). The importance of full assessment of patients’ families cannot be overemphasized, whether in adults or in children (Dyck et al. 1981a; Hagberg and Lyon 1981).
The syndrome of hereditary CMT or peroneal muscular dystrophy covers a heterogeneous group of inherited polyneuropathies (Rautenstrauss 2011) that have a similar clinical phenotype. Charcot–Marie–Tooth disease historically has been divided into the demyelinating or hypertrophic forms (CMT1) in which nerve conduction velocity is markedly and diffusely reduced) and neuronal forms (CMT-2) characterized by normal or slightly slowed nerve conduction velocity) (Shy and Patzko 2011). A third form, intermediate CMT ( X-linked CMT), comprises patients in whom the MCV lies in the range of 25 m/s and 45 m/s. Early-onset genetic neuropathies (vide infra), including those of prenatal development, are incorporated into the forms of CMT-1 and CMT-2.
Two other less frequently inherited groups of polyneuropathies are closely related to CMT: the hereditary sensory neuropathies (HSN), which affects predominantly sensory fibers, and the distal hereditary motor neuropathies (HMN), which display only motor involvement (Rossor et al. 2013). These three disorders are collectively included into the family of CMT by some authors.
Traditional classifications of CMT diseases combine elements of clinicopathological phenotypes, inheritance patterns, motor nerve conduction velocities, and causative genes. Up to the end of 2013, 70 disease genes have been described for CMT (of which 33 were discovered after 2009!) and related disorders. Nonetheless, more than 70 % of patients with CMT, in Western countries, have genetic abnormalities affecting only four genes: PMP22, MPZ, MFN2, and GJB1 (Tazir et al. 2013).
The rapid evolution in gene discovery is attributed to the development and affordability of high-throughput sequencing technologies, has affected diagnosis and management of patients with CMT, and has enabled the design of classifications where genetic data plays a significant role (Rossor et al. 2013).
Clinical and pathological similarities exist between CMT and some hereditary spastic paraplegias with a subset of patients having an overlap condition termed Silver syndrome (Timmerman et al. 2013).
CMT may be the presenting phenotype of mitochondrial disorders. MFN2 and GDAP1 are involved in the fusion and fission of mitochondria, fundamental in regulating mitochondrial transport along the cell. CMT can result from mutations of MFN2 (autosomal dominant CMT-2A, autosomal recessive CMT-2, CMT-5, and CMT-6) and GDAP1 (CMT-2 K, CMT-4A, recessive intermediate CMT, and autosomal dominant CMT CMT-2 K) (Pareyson et al. 2013) (vide infra).
19.2 CMT-1
Traditionally referred to as hypertrophic Charcot–Marie–Tooth disease or hereditary sensory motor neuropathy (HSMN-I), the central features of CMT-1 are marked slowing of nerve conduction velocity with demyelination, remyelination, and onion bulb formation. Nerve conduction studies play a critical role in separating the hypertrophic and the neuronal forms of CMT. Conduction velocities of less than 38 m/s have been proposed to separate CMT-1 from CMT-2 (Harding and Thomas 1980a). Slowing of conduction is seen in asymptomatic or presymptomatic carriers (Nicholson 1991). CSF examination is normal or shows slight elevation of protein.
Different inheritance and molecular-genetic studies within this group of patients indicate that CMT-1 is a genetically heterogeneous syndrome. The most common variant, accounting for 70–80 % of all patients, is now known to be due to a defect in expression of PMP22 protein that is caused by a point mutation or tandem duplication of the PMP22 gene (on chromosome 17p11.2–p12) (Hoogendijk et al. 1993). PMP 22 is an integral membrane protein produced by Schwann cells, where it plays a crucial role in the development and maintenance of compact myelin. A smaller group linked to chromosome 1, classified as CMT-1B (Lebo et al. 1991; Ionasescu et al. 1993), is characterized by defects in the MPZ gene which codes for the peripheral myelin protein P0 (Hayasaka et al. 1993c). Patients with CMT-1B are clinically indistinguishable from patients with CMT-1A. Other cases of autosomal dominant CMT-1 (CMT-1C, CMT-ID, CMT-IF, and CMT1 plus retinal degeneration) are caused by LITAF, EGR2 (which influences the transcription of other myelin protein genes), NEFL, and FBLN5 gene defects (Rossor et al. 2013). The CMT-1A and CMT-1B groups demonstrate autosomal dominant inheritance with age-dependent (eventually 100 %) but variably severe penetrance. In Western Europe, North America, and Japan, the dominant forms of CMT-1 are by far the most frequent (Tazir et al. 2013). About 20 % of cases are sporadic, and although segregation analysis had previously suggested that most were recessive (Harding and Thomas 1980c), recent genetic studies indicate that most sporadic cases are caused by new mutation of the CMT-1A type (Hoogendijk et al. 1992; Gabreels-Festen et al. 1993). Autosomal recessive forms are classified as CMT-4 (see Charcot–Marie–Tooth disease type 4).
19.2.1 Clinical Manifestations
While individual cases of CMT-1A and CMT-1B are not clinically distinguishable, CMT-1B is typically more severe (Dyck et al. 1993). Symptoms usually begin in the first or second decade although nerve conduction is abnormal in early life. Onset in the 3rd–5th and rarely up to seventh decade is well described (Harding and Thomas 1980a). The usual presenting manifestations are skeletal deformity, distal leg muscle atrophy, weakness, or indirectly through findings detected while undergoing evaluation for non-neurological symptoms (Dyck et al. 1993). Pes cavus, hammertoes, and scoliosis may be present as early manifestations in childhood and serve as important clues to the diagnosis of a familial neuropathy. Symptoms can be so mild and insidious that some patients never present to a physician for disease-related complaints. Visible and palpable hypertrophy of peripheral nerves, especially the greater auricular nerve, can be found in approximately a quarter of patients. Weakness and atrophy are often restricted to the distal legs, even after many years of illness, with about half of patients progressing to distal hand muscle weakness. Reflexes are typically diminished or absent in a distal to proximal gradient. Sensory loss involves all modalities but is usually less prominent than motor symptoms, joint destruction, or sensory ataxia being uncommon. Essential tremor is seen in a third of CMT-1 patients and its presence defines the “Roussy–Lévy” variant, but these patients are not genetically distinct from those with other CMT-1 variants. The disease progresses very slowly over a patient’s lifetime and does not influence most individuals’ longevity; only a minority eventually become wheelchair dependent. A small subset of patients with dominantly inherited CMT-1 present in early childhood or even at birth, some progressing very slowly, but others reaching a debilitated state (Ouvrier et al. 1987; Vanasse and Dubowitz 1981; Hagberg and Lyon 1981).
Medication-induced (vincristine, metronidazole, nitrous oxide, nitrofurantoin, etc.) exacerbation of neuropathy in CMT is well known (Weimer and Podwall 2006). In CMT patients, acute initiation of weakness, including a Guillain–Barré-like pattern, may occur. In ten previously unsuspected and undiagnosed CMT patients, nerve deficit developed following the administration of pharmaceuticals as the initial manifestation of the genetic disease.
19.2.2 Pathology
19.2.2.1 Role of Biopsy
Given that a familial neuropathy is often clinically suspected, that examination of even “normal” family members in CMT-1 will often disclose affected individuals, and that nerve conduction studies can distinguish between hypertrophic and neuronal variants, there is little reason to biopsy patients in this setting. Some of the cases referred for biopsy in our institutions had been suspected to represent CIDP because of atypical features. For the most part, however, the diagnosis of CMT-1 was strongly suspected clinically, but perhaps because there is no specific therapy to offer, the referring clinicians felt the need to prove their suspicion and make every effort to rule out a treatable neuropathy (especially CIDP) before offering genetic counseling. Even so, recent advances in molecular biology permit noninvasive diagnosis of CMT-1A with greater specificity than biopsy (Hoogendijk et al. 1992; Gabreels-Festen et al. 1993), and rapid progress has been made in the genetic characterization of the other forms of CMT (Rossor et al. 2013).
In the setting of a long history consistent with CMT, but a recent rapid worsening, the possibility of a superimposed diagnosis, such as CIDP or paraproteinemic neuropathy, should be considered. Authors observing such a co-occurrence have suggested an increased risk for CIDP or paraproteinemic neuropathy with CMT-1 (Gregory et al. 1993; Dyck et al. 1982). Although clinical acumen is more important than biopsy for the identification of such patients, histological examination may provide useful confirmatory evidence. Sural nerve biopsy may be diagnostic of familial neuropathy even if a patient is clinically and electrophysiologically normal (Dyck et al. 1983).
In infants and young children, where the spectrum of clinical and histological findings is wide, biopsy may provide useful prognostic and inheritance information. Infantile or childhood-onset CIDP may be clinically indistinguishable from inherited neuropathies, but is treatable and should not be overlooked (Sladky et al. 1986). Although there is disagreement (Hagberg and Lyon 1981), the literature suggests that biopsy can reliably separate CMT-1 and CMT-3, a distinction with prognostic and genetic implications (vide infra). The light-microscopic features are also indistinguishable, but an ultrastructural finding that suggests the diagnosis of the myelin P0 (MPZ) mutation (CMT-1B) is the presence of uncompacted myelin.
19.2.2.2 Light Microscopy
The literature contains several large biopsy series, and our experience is entirely congruent with the information these present (Behse and Buchthal 1977; Gabreels-Festen et al. 1992b; Madrid et al. 1977; Gherardi et al. 1983; Ouvrier et al. 1987; Low et al. 1978).
An increase in the size of nerve fascicles, sometimes severalfold, is often readily apparent in advanced CMT-1, reflecting the additive accumulation of diffuse onion bulb formations, edema, extracellular matrix, and collagen in the endoneurium (Behse et al. 1974). Histochemical stains often indicate that the endoneurial interstitial “edema” accumulation has a mucoid component. Proliferation of Schwann cells and fibroblasts results in increased endoneurial cellularity. The fascicle-to-fascicle distribution of pathology is relatively homogenous. Variable amounts of chronic inflammation can be found in some cases (Thomas et al. 1997). Many thinly myelinated fibers are seen. Increased myelin spiral length in relation to axon diameter, which has been interpreted as evidence of axonal atrophy, possibly reflects overexpression of PMP22, leading to myelin thickening before demyelination occurs.
A reduction in myelinated fiber (MF) numbers is evident and varies in severity from moderate to subtotal, correlating with duration and severity of disease. Both large and small MFs are depleted (Fig. 3.1c), and a unimodal size-frequency histogram is characteristic (Gabreels-Festen and Gabreels 1993). Unmyelinated fiber (UF) densities are usually normal, and sometimes even increased (Tome et al. 1979). Regenerating clusters are ordinarily found in numbers no greater than controls, but we have seen them feature prominently in some cases which were otherwise typical of CMT-1 (see “Intermediate CMT” below).
Repeated episodes of primary segmental demyelination and remyelination lead to the formation of onion bulbs, a term that refers to imbricated layers of supernumerary Schwann cell processes forming rings around individual axons, an appearance which can be confidently detected only in cross sections. Although hypertrophic neuropathy is a prominent feature of several of the genetically determined neuropathies, onion bulbs can also occur in CIDP and, to a lesser extent, in diabetic neuropathy and other acquired polyneuropathies. The hallmark of familial hypertrophic neuropathy is the presence of onion bulbs (OBs) involving a substantial fraction (30–100 %) (Gherardi et al. 1983; Behse et al. 1974; Low et al. 1978) of the visible myelinated and demyelinated axons, distributed diffusely among the nerve fascicles (Figs. 19.1 and 19.2). Onion bulbs can be visualized in paraffin-embedded material using H&E sections (Fig. 19.1a), an appearance confirmed by collagen IV staining of Schwann cell basal laminae (Fig. 19.1b). Onion bulbs may have a central myelinated or demyelinated axon (arrows, Fig. 19.1c), no central axon, or several axons as shown by neurofilament immunoreactivity. Paraffin-embedded thick sections do not reveal these structures as well as as semithin toluidine blue (Figs. 19.1d and 19.2a–d) and especially ultrathin EM sections (Fig. 19.3). Naked axons may be present, more frequently in children (arrow, Fig. 19.2d). Myelin debris is rarely seen, especially in adults (Fig. 19.2d). While myelin sheaths can be inappropriately thin indicating remyelination, they may also appear normal or even hypermyelinated relative to axon diameter (arrow, Fig. 19.2b; arrowhead, Fig. 19.2c) and may show focal redundant loops (Dyck et al. 1970; Gabreels-Festen et al. 1992b). Small myelinated fibers in particular may demonstrate inappropriately thick myelin for axonal diameter, perhaps due to axonal atrophy (arrowhead, Figs. 19.2c and 19.3).
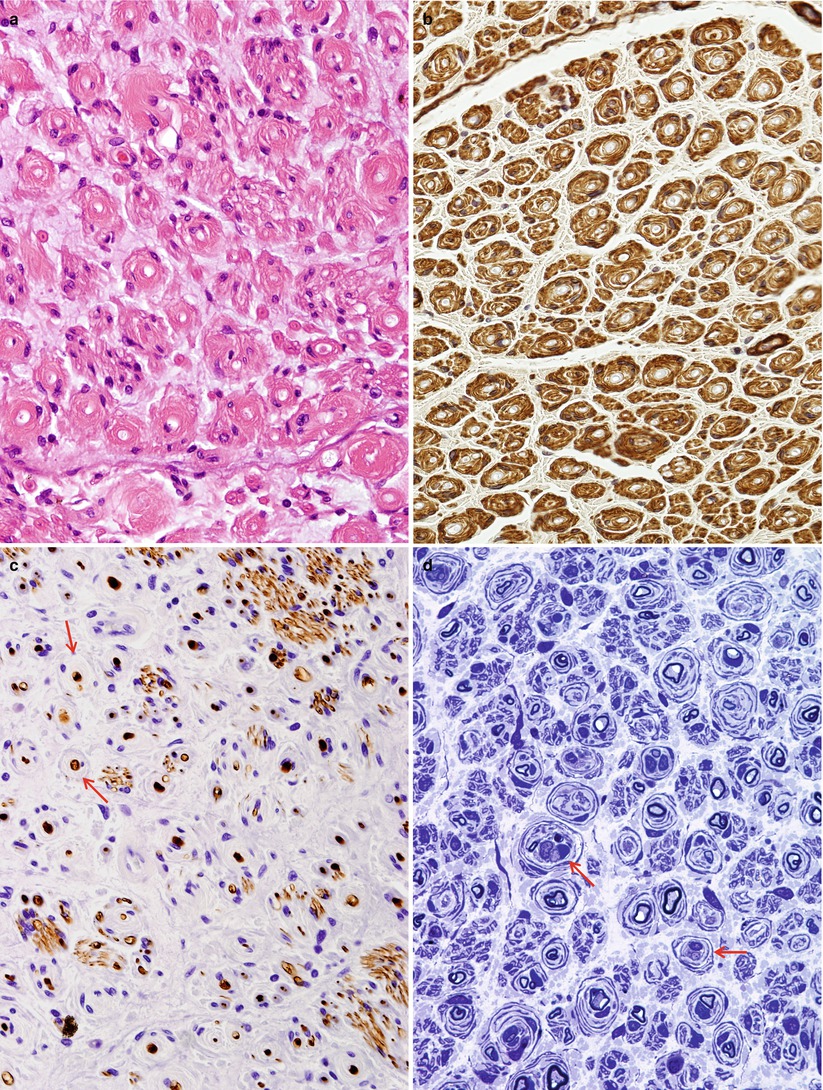
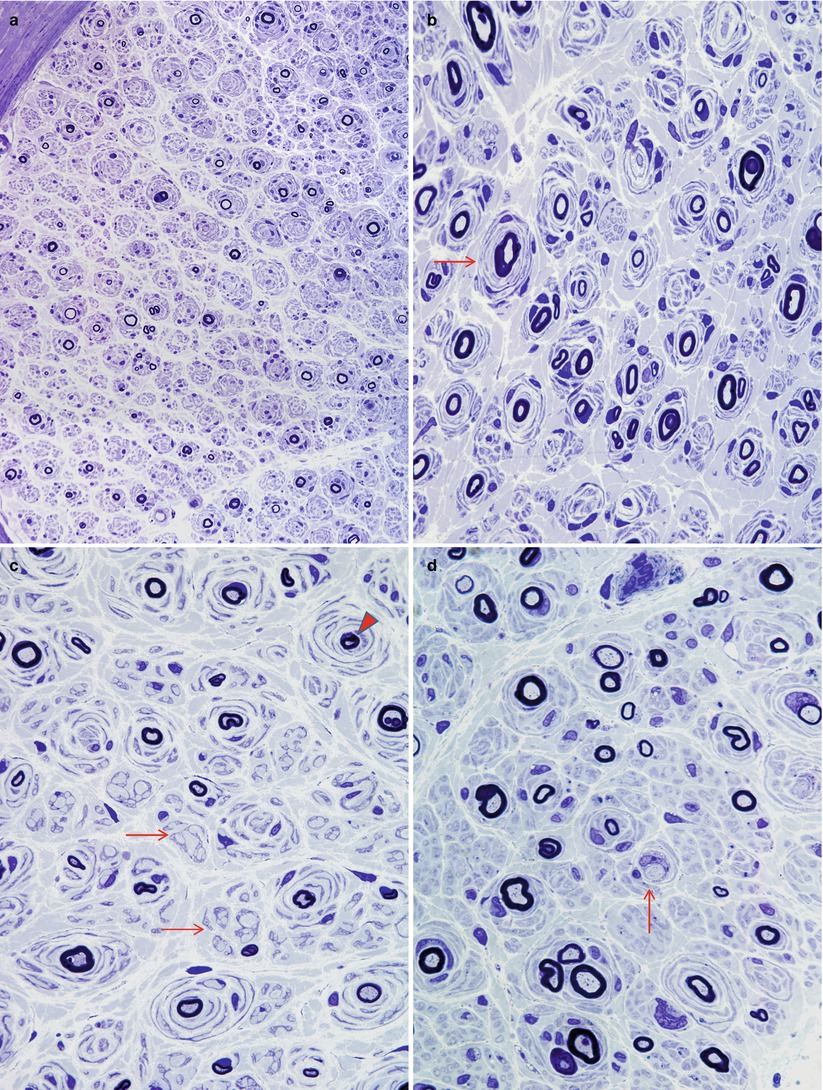
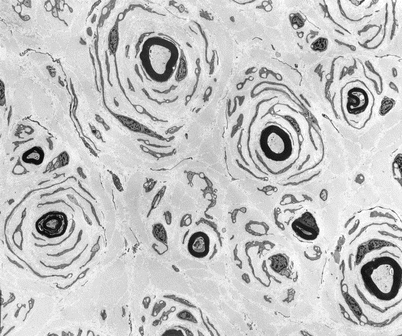
Fig. 19.1
Onion-bulb neuropathy: characteristic histopathology. (a, b) Onion bulbs consist of prominent concentric rings of Schwann cell processes surrounding a central pale axon by H&E (a) and collagen IV immunohistochemistry (b). (c) Single axons form the center of most onion bulbs (arrows), although some have several axons or lack axons altogether (neurofilament immunohistochemistry). (d) Plastic sections show onion bulbs with thinned or absent (arrows) myelin sheaths (Magnification: paraffin, a, H&E, 400×; b, collagen IV, 400×; c, neurofilament, 400×; d, 1 μm thick plastic section 1,000×)
Fig. 19.2
CMT-1: spectrum of onion bulb morphology from different cases is shown. Most axons have attenuated myelin (a–d), while some appear relatively hypermyelinated (arrow, b; arrowhead, c). (c) The delicate processes between onion bulbs (arrows) represent denervated Remak bundles. (d) Naked axons are also noted (arrow) (Magnification: 1 μm thick plastic section a, 400×; b–d, 1,000×)
Fig. 19.3
CMT-1: classic ultrastructural appearance of onion bulbs (Magnification 2,250×)
Evolution of the histological picture from childhood into adult life has been studied by several authors (Gabreels-Festen et al. 1992b; Ouvrier et al. 1987; Meier et al. 1976). Children have fewer and smaller OBs, sometimes almost none, while naked or actively demyelinating axons are more prominent. In one study 2.7–9.2 % of MFs were demyelinated and 0–1.7 % were actively demyelinating in patients below the age of 10, whereas 0–2 % and 0–0.2 % respectively showed these changes in patients above the age of 10 (Gabreels-Festen et al. 1992b). In children debris-filled cells remain unusual but are more common than in adults. The finding of a fair number of naked axons with only a few cells containing myelin debris suggests delayed remyelination. With increasing age the active segmental myelin changes become less frequent, and OBs increase in number and size. By the age of 6, onion bulbs are usually well developed (Gabreels-Festen et al. 1992b). Loss of MFs is also age related, although even at early stages there is a significant diminution in their numbers. As with adults, degenerating axons are generally not seen. However, nonspecific axonal alterations, particularly atrophy, can be present. Denervated Schwann cell subunits and collagen pockets may be seen more often than in normals, indicating unmyelinated fiber disease, but UF density is usually maintained or even increased (Behse and Buchthal 1977; Low et al. 1978).
19.2.2.3 Electron Microscopy
Onion Bulb Formations
Onion bulb formations are a nonspecific but nevertheless significant finding on nerve biopsy. They are discussed here in detail because they comprise a central part of the histological picture in CMT-1. However, there are no morphologic features of a single onion bulb which allow specific diagnosis. Helpful clues may be obtained from the pattern of distribution of OBs within the nerve fascicle and from other findings in the nerve biopsy.
Onion bulbs are easily detected by light microscopy on semithin sections, particularly under oil immersion, but they are best defined with the electron microscope. A typical formation consists of a myelinated axon surrounded by concentric layers of Schwann cell processes; these can be plump, thin and elongated, or tightly packed stacks (Figs. 19.3 and 19.4). The identity of the cellular profiles is established by the finding of a surrounding basal lamina. Collagen fibers run longitudinally and separate the individual OB layers. Numerous variations on this theme can be seen. An individual axon may be denuded, thinly myelinated, or hypermyelinated. An OB forming around what appears to be a large unmyelinated axon (Fig. 19.4c) likely represents a previously myelinated fiber that has become atrophic with the loss of its myelin sheath. Rarely, more than one myelinated axon is seen in the center, representing a regenerating cluster within an onion bulb.
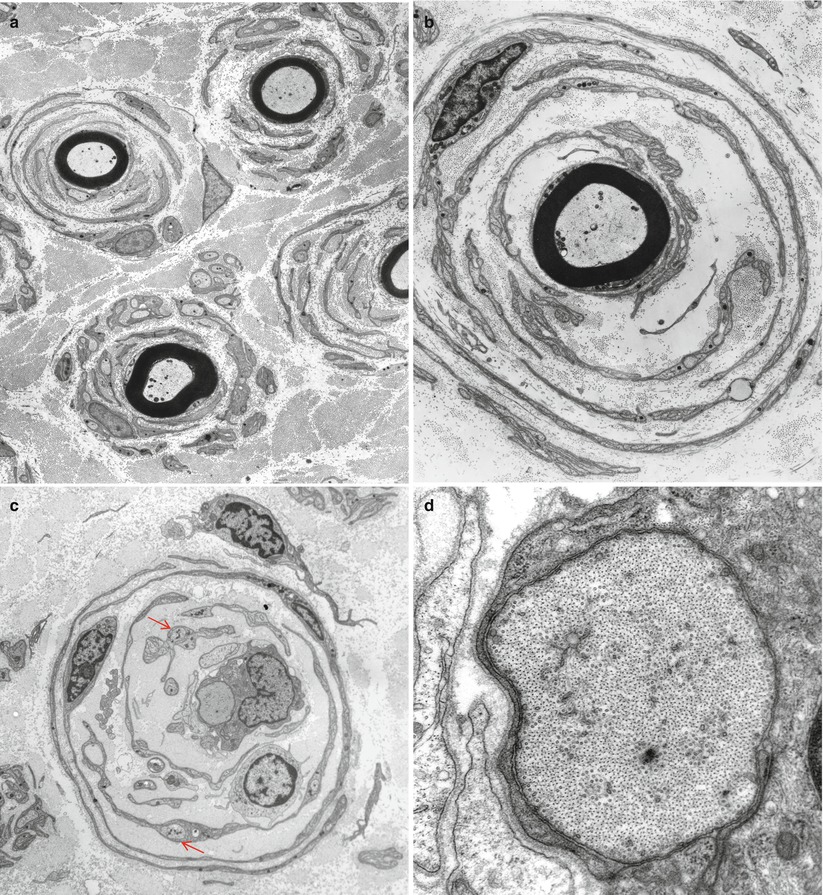
Fig. 19.4
CMT-1: (a, b) classic onion bulbs consist of concentric imbricated Schwann cell processes alternating with collagen fibers. (c, d) Note central naked axon in (c) which is also shown at higher magnification in (d). The demyelinated axon contains increased numbers and density of neurofilaments and microtubules, reminiscent of concentrated intra-axonal organelles seen at nodes of Ranvier. A few small unmyelinated axons are seen in surrounding Schwann cell processes (arrows, c) (Magnification: a, 2,240×; b, 3,300×; c, 5,000; d, 40,000×)
Unmyelinated fibers are frequently incorporated into the circumferential lamellae of an OB (arrows, Fig. 19.4c). These UFs may be tiny sprouts arising from the demyelinated axon as part of the regenerative response after segmental demyelination or neighboring UFs which have gravitated into the onion bulb. Ochoa (1978) has observed that the outermost lamellae of OB formations often have the configuration of denervated Schwann cell bands and believes that neighboring UFs can make a significant contribution to OB formation. A “pseudo”-onion bulb is formed when a myelinated axon is surrounded by several unmyelinated axons and their accompanying Schwann cells as part of a regenerating cluster (Fig. 7.13). Misinterpretation of these formations as true onion bulbs may lead to the false impression that a hypertrophic neuropathy is present.
Typically there are 2–5 concentric layers of Schwann cell processes (Gherardi et al. 1983), but we have observed giant OBs with 20 such lamellae (Fig. 9.31). The Schwann cell processes may disappear, leaving behind closely apposed basement membranes. The latter can only be recognized by electron microscopic examination and are infrequently seen in typical cases of CMT-1 (vide infra). Ultimately the axon at the center may disappear, replaced by collagen or Schwann cells, creating a denervated or obsolete onion bulb (Fig. 19.5). The frequency of these structures parallels disease duration.
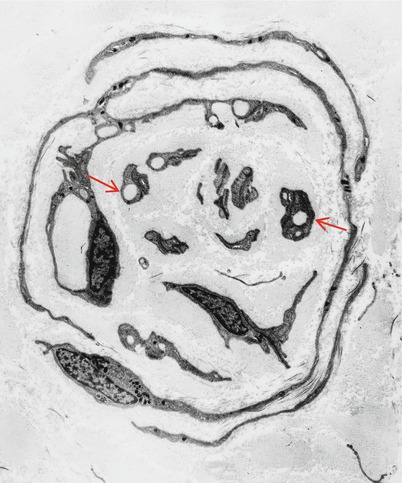
Fig. 19.5
CMT-1, obsolete onion bulb: concentric layers of Schwann cell processes enclose collagen which is arranged longitudinally or circumferentially. Many Schwann cells display collagen pockets (arrows) (Magnification: 3,120×)
Other Ultrastructural Changes in CMT-1
Active demyelination is rarely seen in CMT-1, but when present appears as a focal myelin degeneration or an accumulation of debris in Schwann cells. Debris-filled macrophages are uncommon, but may be more frequently detected in children, where the demyelination is most active (Gabreels-Festen et al. 1992b; Ouvrier et al. 1987; Meier et al. 1976). Madrid et al. (1977) and Vital et al. (1992) have commented on the rare finding of macrophage-mediated myelin stripping in familial hypertrophic neuropathy but we have never seen this, and the illustrative figures may simply represent a scavenger macrophage that has entered the Schwann tube to clear away damaged myelin. Alternatively, this may represent inflammatory demyelination superimposed on familial neuropathy, as described by some authors (Sect. 19.2.4 below). In addition, in mouse models of CMT-1A, CMT-1B, and CMT-1X (mice homo/hemizygously deficient for Cx32), peripheral nerve macrophages and CD8+ T cells have been shown to contribute significantly to the pathogenesis of these genetic disorders (Kobsar et al. 2005; Ip et al. 2006).
Schwann cells in the process of remyelinating recently demyelinated axons are characterized by an increased amount of electron dense cytoplasm and evidence of anabolic activity. A few lamellae of myelin may be deposited, and in the earliest stages of remyelination, these appear uncompacted (Figs. 5.13 and 14.7). Naked or demyelinating axons often seem atrophic and show an increased axonal filament density and a wrinkled axolemma, similar to that described in other demyelinating conditions (Fig. 9.24) (Gabreels-Festen et al. 1992b).
19.2.2.4 Fiber Teasing
In CMT-1 segmental myelin changes, including entirely demyelinated segments, are seen in 30–100 % of teased fibers (Dyck 1966; Dyck et al. 1968, 1993; Behse and Buchthal 1977; Low et al. 1978). Frequent segmental demyelination and remyelination results in shortened (<0.60 mm) internodes. Two thirds of 6 mm segments of teased nerve fibers will show an abnormal internode, rising to 95 % of 8 mm segments, in comparison with 0.5 % of 5 mm segments in normals. Segmental myelin changes cluster on certain fibers, suggestive of secondary demyelination (Dyck et al. 1993). Axonal degeneration is not present significantly more often than in normals.
19.2.2.5 CMT-1 Variants
The description of histological findings provided above applies to the large majority of CMT-1 patients who demonstrate autosomal dominant inheritance. It is not thought that a significant difference exists between the histology of CMT-1A and CMT-1B; however, the large biopsy series of the past were performed before the genetic delineation of the 1A and 1B groups and demonstrated homogenous findings, with the exception of the controversial “intermediate” form (Madrid et al. 1977). CMT-1A is a less severe phenotype (on average) than CMT-1B (Dyck et al. 1989, 1993; Bird et al. 1983), and very scanty evidence suggests that nerve biopsy might parallel this (Bird et al. 1983). However, the dramatic variation of biopsy findings demonstrated in one dominantly inherited kinship makes comparison on the basis of single cases meaningless (van Weerden et al. 1982). In another report two related patients (father and son) with CMT-1B showed very frequent myelin tomaculae and a mutation of myelin P0 protein (Thomas et al. 1994). Such meager evidence makes it difficult to distinguish CMT-1A from CMT-1B histologically.
Some reports of recessive CMT-1 have shown histology similar to that described for dominant CMT, but the available material is scanty (Harding and Thomas 1980b). Early-onset dysmyelinating neuropathies with focally folded myelin or with basal lamina onion bulbs (discussed below with the congenital dysmyelinating neuropathies) are also recessively inherited and may not be distinguishable from the cases described by Harding and Thomas (1980b).
For X-linked CMT there has been a controversy regarding whether the changes are primarily hypertrophic or neuronal. While Rozear et al. (1987) and Nicholson and Nash (1993) describe features suggestive of a hypertrophic neuropathy in a total of 3 biopsies in X-linked kindreds, Hahn argues that the “onion bulb-like” structures seen in two cases in her laboratory were “pseudo”-onion bulbs formed by regenerating clusters and that true onion bulbs are not present (Hahn 1993). X-linked CMT is also genetically heterogeneous (Rossor et al. 2013).
The pathological features seen in a case of homozygous AD CMT-1 were not particularly marked, despite the more severe clinical phenotype (Killian and Kloepfer 1979). In a pedigree demonstrating severe dominantly inherited CMT-1, biopsy showed massive onion bulb formation and nerve enlargement, with hypomyelination, more suggestive of Dejerine–Sottas disease than typical CMT-1. Genetic analysis demonstrated a point mutation in the PMP gene on chromosome 22 rather than the more common PMP-22 duplication (vide infra) (Hoogendijk et al. 1993).
A small number of axons with redundant myelin loops or outfoldings may be infrequently seen in classic CMT-1. However, a few cases are found in the literature, in patients with the childhood or adult onset of a peripheral neuropathy syndrome similar to ordinary CMT-1, which emphasize focal myelin abnormalities, described variously as “unstable myelin” “globules,” “outfoldings,” or “tomaculae.” In some patients biopsy showed numerous focal myelin masses, measuring 10–30 μm in thickness and 15–100 μm in length, found in a paranodal and less often internodal location, typical of those seen in hereditary neuropathy with pressure palsies (Barbieri et al. 1990; Malandrini et al. 1992); these may represent phenotypic variants of HNPP. One interesting case combines a clinical phenotype of CMT-1, a histological picture of typical tomaculous neuropathy, and a documented mutation in the myelin P0 protein (Thomas et al. 1994). A second group of patients combines a clinical picture of CMT-1 with histology revealing frequent focal myelin degeneration into “blebs” or “globules,” quite distinct from the tomaculae of HNPP; this group is discussed further below. Possibly a third group is identified by Gabreels-Festen and Gabreels (1993) as “CMT-1 with focally folded myelin,” discussed with the congenital dysmyelinating neuropathies (Sect. 19.6.5).
19.2.3 Pathogenesis
19.2.3.1 CMT-1A
CMT-1A is defined as an autosomal dominant disease with duplication or point mutation of the PMP22 gene on chromosome 17 (Lupski 1993; Dyck et al. 1993; Rossor et al. 2013), a defect which accounts for the largest group of CMT-1 patients. Furthermore, most sporadic CMT-1 patients have tested positive for this mutation (Hoogendijk et al. 1992; Gabreels-Festen et al. 1993; Mancardi et al. 1994). It is suggested that new mutations arise from errors during gametogenesis due to misalignment of a 17 KB tandem repeat segment which may normally serve to line up the chromosome for recombination events (Lupski 1993). A point mutation in PMP22 is the cause of the mouse Trembler phenotype, long considered a model for CMT-1 (Suter et al. 1992). This notion has been strongly supported by the detection of pedigrees of CMT-1A with a point mutation in the PMP-22 gene, in one case identical to that found in the Trembler mouse (Roa et al. 1993a; Valentijn et al. 1992).
The mechanism by which the genetic defect causes the neuropathy is unknown. A gene dosage effect might be postulated for the majority of patients with CMT-1 who have a duplication. However, a similar phenotype is present in a small number in patients with a point mutation in the PMP22 gene (Roa et al. 1993a; Valentijn et al. 1992). Deletion of a 1.5 MB segment at 17p11.2, the same region duplicated in CMT-1A, has been consistently associated with hereditary neuropathy with pressure palsies (vide infra Sect. 19.3). In one remarkable pedigree, a patient with a point mutation in one PMP-22 gene and a 1.5 MB deletion in the 17p11.2 region of the other chromosome had a CMT-1 phenotype, while two children with only 1.5 MB deletion had the HNPP phenotype, and another child with the point mutation only was clinically unaffected (Roa et al. 1993b). In this family the point mutation appeared to act in a recessive manner, providing a genetic basis for the clinical observation of recessively inherited CMT-1 (Roa et al. 1993b; Harding and Thomas 1980a).
PMP22 has the structure of an integral membrane protein, with 4 highly conserved transmembrane domains, mutations in 3 of which have been shown to cause a dysmyelinating neuropathy in humans or animals (Lupski 1993; Suter et al. 1993). PMP22 shows some structural similarities to proteolipid protein, found only in the CNS, and both are seen only in compact myelin. A possible role in intercellular communication has been postulated; however, the normal function of PMP22, and details of precisely how a defect results in the disease phenotype are unclear (Lupski 1993; Suter et al. 1993).
Prior to the discovery of the CMT-1A mutation, a large body of evidence pointed to a primary axonal cause of the disease, with secondary myelin change (Nukada et al. 1983; Dyck et al. 1974, 1993; Smith et al. 1980). Similar evidence had been used as the basis for the conclusion that secondary demyelination is an important mechanism in a variety of neuropathies, including uremia (Dyck et al. 1971a), Friedreich’s ataxia (Dyck and Lais 1973), paraproteinemia (Ohi et al. 1985), and the permanent axotomy model (Dyck et al. 1981b). Given that overwhelming evidence now suggests a myelin defect, the question of why there was the appearance of a primarily axonal defect is still relevant. Presumably the role of Schwann cells in regulating the axon’s metabolism is more profound than was previously realized, and defects in the first must cause metabolic and physiologic disturbances in the second (Suter et al. 1993) to account for the axonal loss which is a common feature of CMT-1.
19.2.3.2 CMT-1B
The dramatic genetic advances made in sorting out CMT-1A have been applied to the investigation of CMT-1B. The CMT-1B mutation was previously linked to chromosome 1, q21.2-q25, and this coincides with the locus of P0 protein, the major peripheral nerve myelin protein (Hayasaka et al. 1993a). Point mutations in the P0 protein have been found in the large pedigrees of CMT-1B identified a decade earlier by linkage analysis (Hayasaka et al. 1993b, d; Kulkens et al. 1993; Himoro et al. 1993; Su et al. 1993). The 9 kb P0 gene includes 6 exons, of which 2 and 3 are extracellular components, exon 4 is the transmembrane domain, and exons 5 and 6 represent the cytoplasmic domain. The extracellular domain has homologies with the immunoglobulin variable region and has been thought to play a role in the compaction of myelin at the intraperiod line. Disease-associated mutations in the protein’s structure are all located at the extracellular segment of the molecule. Interestingly, a point mutation in P0 protein has been associated with a CMT clinical phenotype and histological features of tomaculous neuropathy (Thomas et al. 1994).
19.2.4 X-Linked CMT
Seven different defects in the gene encoding the gap junction protein connexin-32 have been detected in 8 families with X-linked CMT (Bergoffen et al. 1993b), while unaffected family members and control subjects did not possess the mutations. This type of CMT disease, which is probably the second most common form of the disease (accounting for around 10 % of all CMT patients), is a genetically separate, X-linked condition. More than 250 mutations of the CX32 (connexin 32, gap junction protein β1, GJB1) gene have been found to be the cause of this syndrome (Bergoffen et al. 1993a, b). Males with CMT-X display a more severe clinical, electrophysiological and histopathological disease than heterozygous females, befitting the notion that having two X chromosomes confers some protection. Connexin-32 is a member of a family of transmembrane proteins, the connexins, which assemble to form gap junctions. Immunohistochemistry reveals that these proteins are found in myelinated peripheral nerve fibers at the nodes of Ranvier and at Schmidt–Lanterman incisura (Bergoffen et al. 1993b). The workers identifying this mutation hypothesized that dysfunction of putative intracellular gap junctions at these sites might impair flow of ions and nutrients to the innermost layers of compact myelin and perhaps the axon, resulting in myelin disruption and axonal degeneration (Bergoffen et al. 1993a, b).
The pathological changes are somewhat nonspecific, but just as the condition is often considered “intermediate” between CMT-1 and CMT-2, so too are its nerve conduction velocities and histopathological findings (Sander et al. 1998; Vital et al. 2001). Classic onion bulbs are not very frequent, but “pseudo-onion bulbs” are common. In these formations, a regenerating cluster is centered by a myelinated fiber and surrounded by several unmyelinated axons and their associated Schwann cells. Axon numbers are often relatively preserved in CMT-X, as large myelinated fibers degenerate or atrophy, but regenerative clustering activity provides numerical compensation. As is typical for genetic processes, the histopathological picture appears chronic, with no active axonal loss (i.e., Wallerian degeneration) or active demyelination visible on cross sections, although teased fibers may show some ongoing axonal degeneration.
19.2.5 Differential Diagnosis
When a nerve biopsy shows prominent hypertrophic changes, the most important diagnostic considerations are CIDP and CMT-1. These can usually be distinguished by the pattern of involvement: multifocal in CIDP and diffuse in CMT-1 and definitively by genetic analysis. However, minimal pathological expression of CMT-1 may demonstrate multifocal regions containing onion bulbs interspersed with fibers of normal appearance (Dyck et al. 1983). Prominent inflammatory infiltrates are not typical of classic CMT-1; however, a few epineurial lymphocytes should not be over interpreted as CIDP. Because of the chronicity of the process, macrophages filled with myelin debris are rare in adult CMT-1, and when several are seen in the same section, CIDP should be considered. We do not accept macrophage-mediated myelin stripping as a feature of CMT-1. If evidence is conflicting, consideration should be given to the possibility of CMT with a superimposed inflammatory neuropathy, which may be steroid responsive (Dyck et al. 1982; Bird and Sladky 1991; Vital et al. 1990; Crawford and Griffin 1991). Gabreels-Festen et al. (1993) have considered this issue, and a detailed discussion is provided in Chap. 9. PMP-22 expression in nerve biopsy material can distinguish between CMT-1A and other demyelinating neuropathies, but there is an overlap between the two groups (Yoshikawa et al. 1994).
In an infant with a dysmyelinating neuropathy, the differential diagnostic possibilities include CMT-1, Dejerine–Sottas syndrome, the other congenital dysmyelinating neuropathies (vide infra), and CIDP. An infantile or early childhood neuropathy progressing for many years can be due to CIDP, and nerve biopsy may be the only means of making the diagnosis (Sladky et al. 1986), although nerve conduction studies are very helpful. In the absence of a family history of dominant inheritance, sporadic infantile CMT-1 is distinguished from Dejerine–Sottas in that the latter shows very prominent hypomyelination and demyelination with a g-ratio >0.7, few or no axons over 6–8 μm in diameter on the myelinated fiber histogram, and prominent (>50 % of myelinated fibers) onion bulb formations at a young age (Ouvrier et al. 1987). The distinction is important because prognosis is better for infantile onset CMT-1 than for Dejerine–Sottas syndrome (Vanasse and Dubowitz 1981; Ouvrier et al. 1987). Moreover, nerve biopsy might suggest the mode of transmission, i.e., dominant (CMT-1) or recessive (Dejerine–Sottas). The available literature suggests that biopsies revealing numerous basement membrane onion bulbs or focally folded myelin are more likely to be recessive (vide infra). Uniformly thin myelination is not specific for Dejerine–Sottas syndrome. We have examined CIDP biopsies in adults with a picture of uniformly distributed, strikingly thinly myelinated axons in an adult, and a similar appearance may be present in metachromatic leukodystrophy (Bardosi et al. 1987). Nerve biopsy features typical of Dejerine–Sottas syndrome were seen in a patient with a point mutation in the PMP gene who had dominant inheritance of a particularly severe CMT-1 phenotype (Hoogendijk et al. 1993) and in a patient with homozygous inheritance of dominant CMT-2 (Sghirlanzoni et al. 1992).
CMT-1 can show dramatic histological variability. In one CMT-1 kinship in which 5 biopsies were performed, one showed classic OBs, 2 showed thinly myelinated axons, and 2 showed only axonal loss (van Weerden et al. 1982).
19.3 Hereditary Neuropathy with Pressure Palsies
19.3.1 Clinical Manifestations
Hereditary neuropathy with pressure palsies (HNPP) is an increasingly recognized entity that has emerged in recent decades. Patients experience recurrent bouts of mononeuropathy, sometimes precipitated by trivial injury to the affected nerve and usually resolving over hours to weeks. Painless peroneal, radial, or ulnar palsies are most common (Meier and Moll 1982; Verhagen et al. 1993; Earl et al. 1964), and a mononeuritis multiplex-like picture may occur (Behse et al. 1972). Brachial plexus involvement is not infrequent, accounting for 8 % of episodes in one review (Meier and Moll 1982), while cranial nerves are only rarely affected. A mild polyneuropathy may be superimposed. Uncommonly, weakness in the territory of a single nerve can be slowly progressive but may remit even after many years if close attention is paid to avoidance of pressure or traction (Earl et al. 1964, case 19-1). Although most patients identify the second or third decades as the time of disease onset, focal paralysis may be present at birth. Alternatively, the diagnosis can be delayed as long as the seventh decade if the recurrent nature and family history of such symptoms are not appreciated (Meier and Moll 1982). The inheritance pattern is autosomal dominant, with complete or nearly complete penetrance but variable disease severity (Verhagen et al. 1993). Genetic studies have shown point mutation in PMP22 or deletions affecting the region of chromosome 17 harboring the PMP22 gene (Chance et al. 1992, 1993) in 85 % of patients. Therefore, HNPP can be caused by deletion of the gene PMP22 while duplication of PMP22 causes Charcot–Marie–Tooth disease type 1A, and point mutation in PMP22 may result in either HNPP or CMT1A.
Rarely, patients with the typical histological findings of HNPP seem to suffer a slowly progressive, distally predominant sensorimotor polyneuropathy (Madrid and Bradley 1975; Malandrini et al. 1992; Barbieri et al. 1990; Pellissier et al. 1987) similar to the Charcot–Marie–Tooth syndrome. It is unclear if this is a phenotypic variant, a spurious consequence of insufficient detailed electrophysiological testing (Felice et al. 1994), or an entirely different disease, perhaps CMT-1B (Thomas et al. 1994). The pediatric cases of HNPP reported by Gabreels-Festen and colleagues (1992a) often had a chronic progressive neuropathy, while a history of recurrent focal palsies could be found in some of the affected older relatives, suggesting that HNPP can manifest as a chronic progressive neuropathy in early life, with the typical focal palsies coming later. Other pedigrees exist in which some affected individuals showed recurrent focal palsies while others had only a chronic neuropathy (Leblhuber et al. 1991).
Electrophysiological testing reveals a diffuse abnormality of peripheral nerves, with mild-moderate slowing of conduction, worse distally, even in clinically unaffected nerves. Sites of nerve compression are particularly likely to be affected. EMG reveals neurogenic changes, usually chronic, indicating that axonal degeneration does occur (Behse et al. 1972). CSF examination is unremarkable.
19.3.2 Pathology
19.3.2.1 General Considerations
Although Behse and colleagues (1972) were the first to describe the classic pathology, it was Madrid and Bradley (1975) who coined the term “Tomaculous Neuropathy.” At least two of the four patients described in this report suffered from recurrent pressure palsies, and the term “tomaculous neuropathy” has often been used synonymously with HNPP. However, myelin “tomaculae” are seen in other settings such as the dysmyelinating neuropathies and some patients with a Charcot–Marie–Tooth syndrome (vide supra). Thus, the term “tomaculous neuropathy” should be used in a descriptive sense only. The histological features of HNPP are well described (Madrid and Bradley 1975; Behse et al. 1972; Verhagen et al. 1993; Pellissier et al. 1987; Yoshikawa and Dyck 1991; Drac 1989), and the pathology does not appear to differ significantly in children (Gabreels-Festen et al. 1992a). We have examined six cases.
19.3.2.2 Light Microscopy
Light microscopic examination usually reveals a normal myelinated fiber density and mild nonspecific changes such as endoneurial fibrosis or vascular hyalinization, although occasionally individual markedly enlarged axons can be seen in H&E-stained paraffin sections (Fig. 19.6a, b). The striking abnormality is the presence of many fibers, large and small myelinated axons alike, with a myelin sheath which may be thickened circumferentially or eccentrically or may show prominent focal outfoldings and infoldings (Fig. 19.6c–f). Such changes are seen in <1 % to 10 % of axons in cross sections. The most dramatic circumferentially hypermyelinated fibers often show a “jelly roll” appearance (Figs. 19.6c, e, f and 19.7). Axons associated with such configurations often seem to be constricted, with an abnormally dense axoplasm. In the same specimen, some axons have inappropriately thin sheaths, suggesting demyelination and remyelination.
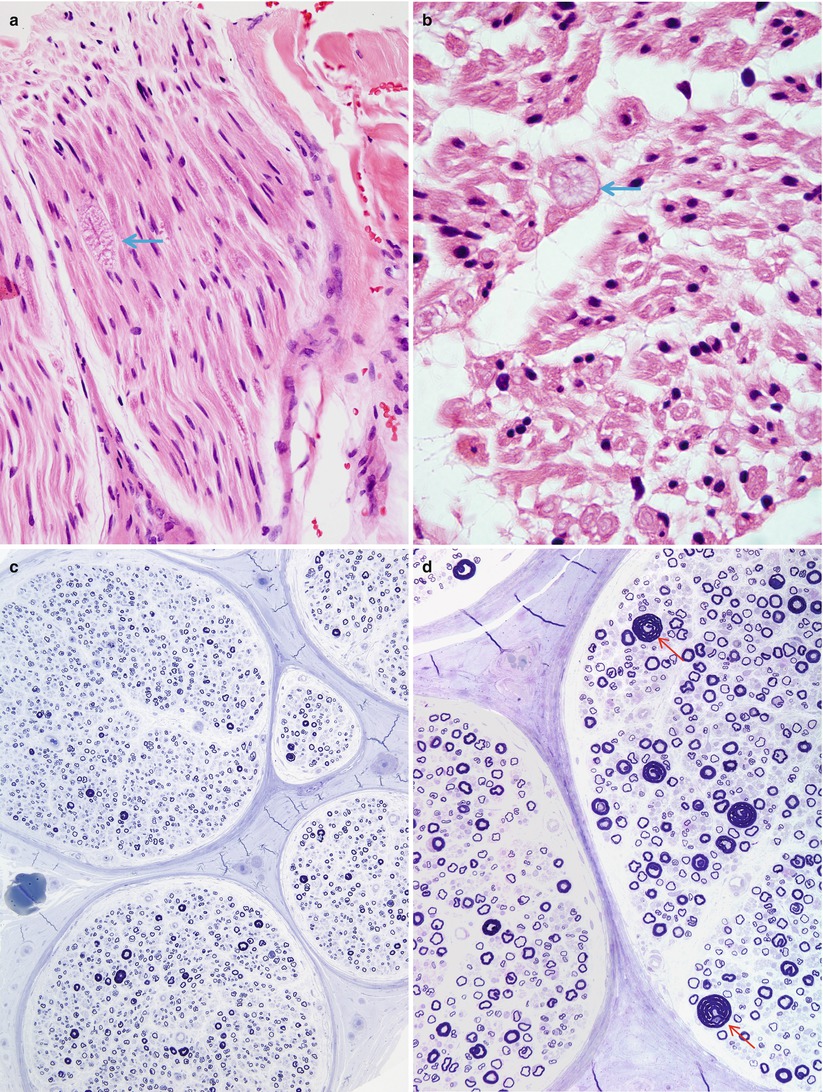
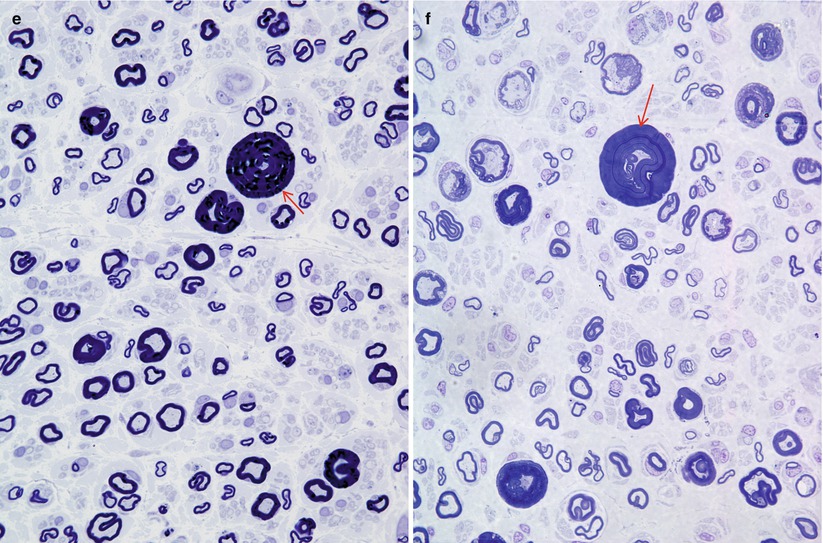
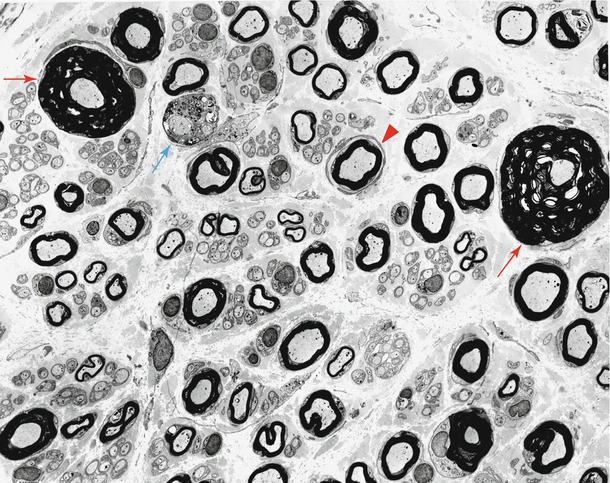
Fig. 19.6
HNPP: (a, b) H&E-stained nerve shows a single tomaculum visible in this fascicle in longitudinal (arrow, a) and cross section (arrow, b). (c–f) Note the relative preservation of axons (c), wide variation of myelin thickness relative to axon diameter and scattered “jelly rolls” (arrows, d–f) (Magnification: a, b, 600×, H&E paraffin; c, 200×,1 μm thick plastic section; d, 400×,1 μm thick plastic section; e, f, 1,000×)
Fig. 19.7
HNPP: low-power view showing the spectrum of findings including “jelly rolls” (red arrow), active demyelination (blue arrow), and rudimentary onion-bulb formation (arrowhead) (1,740×)
In HNPP, intramuscular nerves may show focal myelin swellings (Oda et al. 1990), but these should be distinguished from axonal swellings, which are a common and nonspecific abnormality (Alderson 1992).
Most often myelinated fiber counts are normal, and active axonal degeneration is rare. In a total of 29 cases from three large series (Pellissier et al. 1987; Verhagen et al. 1993; Behse et al. 1972), only 3 showed a mild-moderate reduction in axon numbers. However, the rare observation of degenerating axons and regenerating clusters suggests that axons are not entirely spared in HNPP (Verhagen et al. 1993). The largest diameter fibers are reduced in number relative to controls (Behse et al. 1972; Pellissier et al. 1987; Verhagen et al. 1993). Unmyelinated fibers seemed to be entirely spared in one study (Behse et al. 1972) but showed a shift in diameter towards the small end of the spectrum in another (Pellissier et al. 1987). These changes in the myelinated and unmyelinated axon diameter-frequency histograms might be interpreted as due to axonal atrophy (which may be secondary to myelin abnormalities in myelinated fibers) or as a consequence of degeneration and regeneration.
19.3.2.3 Electron Microscopy
The true nature of myelin tomaculae is revealed best with electron microscopy (Fig. 19.7). All sizes of axons are involved, even individual fibers in a regenerating cluster. Madrid and Bradley (1975) provide a detailed exposition of the various alterations. The most common formation is a double-folded redundant myelin loop that wraps around the axon to varying degrees, forming up to 3 or 4 complete tightly packed turns (Fig. 19.8a) but can be mimicked by compaction of a spiraled redundant myelin loop (Fig. 19.8b). Most often the redundant loops wrap around the outside of the fiber, but they may turn inwards and wrap around the axon internally (Madrid and Bradley 1975). Infolded or outfolded redundant myelin loops that are not adherent to the contour of the axon are common (Fig. 19.9). The existence of true circumferential hypermyelination, defined as a sheath that is normally constructed except for a pathological increase in the number of myelin lamellae, remains to be proven (Fig. 19.8c) (Meier and Moll 1982; Madrid and Bradley 1975; Behse et al. 1972). Whereas normally there are less than 200 myelin lamellae around even the largest of sural nerve axons, up to 480 such lamellae were described in one report (Meier and Moll 1982). However, Yoshikawa and Dyck (1991) feel that close examination of “hypermyelinated” fibers (such as Fig. 19.8b) will show that the increase in myelin lamellae is due to either folding of internodal myelin in a circumferential or longitudinal direction or a result of relative axonal atrophy. In the cases we have examined the fragility of massive myelin tomaculae made it impossible to find a grossly hypermyelinated axon (Fig. 19.8c) whose lamellae were sufficiently well preserved to resolve this issue.
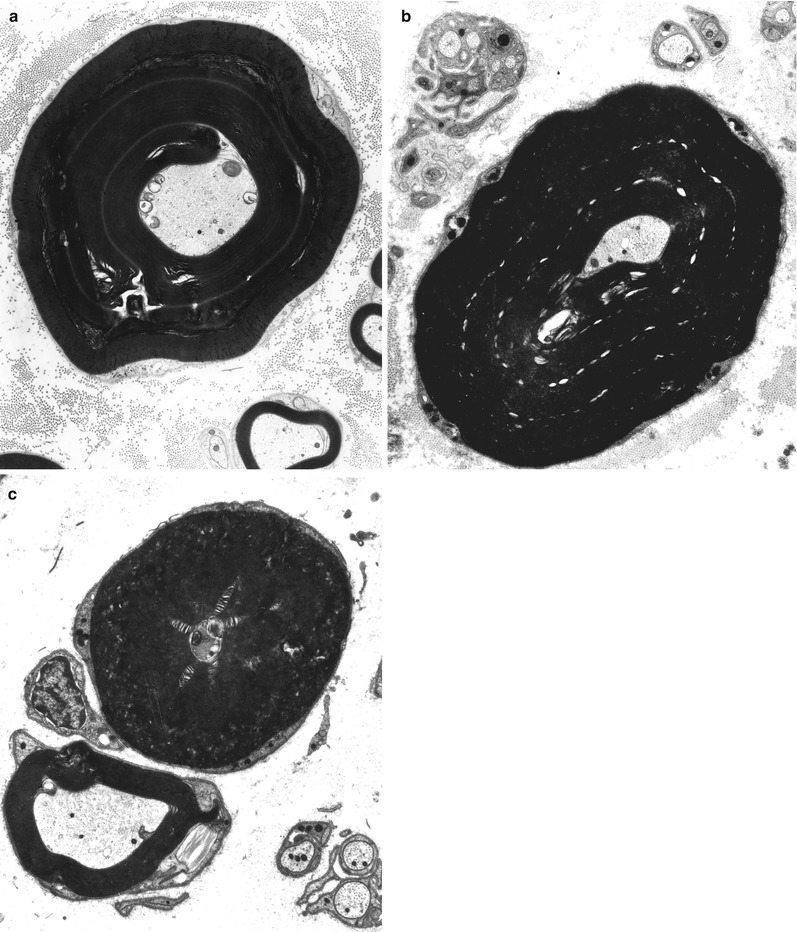
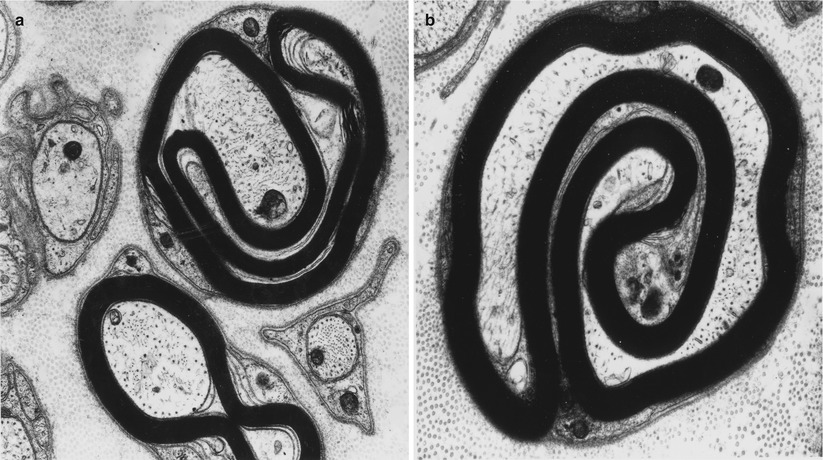
Fig. 19.8
HNPP: “hypermyelination,” spurious “hypermyelinated” fiber formed by compaction of spiraled redundant myelin loop is shown in (a, b). Ultrastructural examination failed to resolve the nature of increased myelin thickness in (c) (a, 5,000×; b, 6,160×; c, 6,840×)
Fig. 19.9
HNPP: redundant outer and inner loops of attenuated myelin. In (b) note the unusual axonal configuration, almost divided by the myelin infolding (a ×15,600, b ×18,000)
Other myelin alterations may be seen. In hypermyelinated fibers, the sheath often appears somewhat ragged, and there may be frank myelin degeneration with debris and denuded axons (blue arrow, Fig. 19.7). Onion bulb formations may be seen but are not usually a dominant feature (arrowhead, Fig. 19.7). At nodes of Ranvier, myelin from one side may seem to overgrow the node and bridge over into the adjacent internode, so-called transnodal myelination. Some workers were able to demonstrate two Schwann cell nuclei per internode (Madrid and Bradley 1975). Uncompacted myelin has been described in HNPP (Yoshikawa and Dyck 1991; Jacobs and Gregory 1991; Hall 1994) and may appear as enlarged Schmidt–Lanterman incisura (Debruyne et al. 1980). This seems to be an uncommon finding that requires an extensive search, for the majority of ultrastructural descriptions of HNPP do not comment on uncompacted myelin, even when specifically looking for it (Verhagen et al. 1993). We have not observed convincing examples of this alteration in our material. The uncompacted lamellae are said to consistently be found on the axonal aspect of the myelin sheath and were seen on normal and on demyelinated/remyelinated internodes alike (Yoshikawa and Dyck 1991; Jacobs and Gregory 1991).
The hyperplastic myelin sheath seems to be unduly fragile and is often seen to be disrupted in a variety of ways, including focal intramyelinic vacuoles, myelin splitting, granular or vesicular myelin disintegration, and at times complete degeneration. This does not occur to the extent seen in the “Neuropathies with Unstable Myelin ” (Sect. 19.2.5, above). Focal collections of degenerating myelin are often located between an intact outer myelin layer and the axon.
Some authors have used the size of myelin tomaculae as a criterion for distinguishing the myelin swellings in HNPP from those of other tomaculous neuropathies. In HNPP tomaculae can be up to 40 μm in width (axon + thickened myelin), with a longitudinal extension of 30–300 μm (Meier and Moll 1982; Behse et al. 1972 ).
At times, an infolded myelin loop or circumferentially thickened myelin may appear to constrict, even obliterate, of the axon within. The axon may show increased filament and tubule density. Serial cross sections of a single fiber indicate that proximal and distal to the area of constriction the axon can regain its normal diameter (Behse et al. 1972). This may be an example of the postulated “demyelinative internal strangulation” mechanism of axonal degeneration discussed by Dyck et al. (1993).
19.3.2.4 Teased Fiber Studies
Tomaculous neuropathies are dramatically demonstrated upon examination of teased fibers (Fig. 19.10). The focal nature of the myelin swellings is best appreciated, as is the distribution, paranodal or internodal (Drac 1989). Typically 25–50 % but anywhere from 5 to 82 % of internodes show focal myelin swellings (Madrid and Bradley 1975; Behse et al. 1972; Verhagen et al. 1993; Pellissier et al. 1987; Drac 1989). Several tomaculae can be seen in one internode, up to 6 in one study (Madrid and Bradley 1975). Teased fibers will also reveal evidence of segmental demyelination and remyelination.
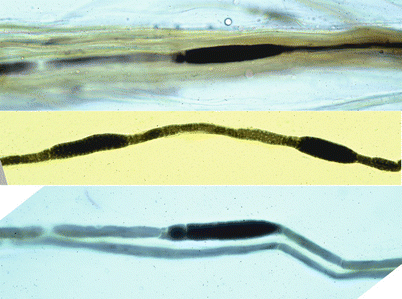
Fig. 19.10
HNPP: teased nerve fiber preparation. This shows characteristic finding of tomaculae and remyelinated internodes
19.3.2.5 HNPP without Myelin Tomaculae
Not all patients with HNPP actually show myelin tomaculae (Castaigne et al. 1976; Earl et al. 1964; Mayer and Garcia-Mullin 1968; Roos and Thygesen 1972; Pellissier et al. 1987; Grossiord et al. 1973; Lhermitte et al. 1973; Attal et al. 1975), although some of the cases and pedigrees described are less than convincing, and electron microscopy, fiber teasing, and semithin toluidine blue-stained sections were often not performed in these reports. The findings include entirely normal biopsies, diffuse loss of myelinated fibers, or nonspecific segmental myelin changes. Absence of the tomaculous change and emergence of fiber loss may indicate a late stage in the evolution of HNPP (Windebank 1993), but many biopsied patients showing tomaculous change have had symptoms for decades (case 19.1).
19.3.3 Pathogenesis
Madrid and Bradley (1975) have discussed the mechanisms that might form the myelin swellings of HNPP, including infoldings, outfoldings, and splitting of the mesaxon. These authors pointed out that similar findings are seen during myelinogenesis but that ordinarily myelin remodeling produces the normal smoothly contoured sheath. The morphologic findings of HNPP are not disease specific, but can be seen, albeit less dramatically, in other demyelinating diseases. A reasonable postulate is that there is a defect in either the ability of the axon to send regulating signals to the Schwann cell or a defect in the Schwann cell’s ability to interpret signals from the axon.
Yoshikawa and Dyck (1991) observed uncompacted myelin in HNPP and found evidence that this change could precede segmental demyelination. The suggestion was made that the neuropathy might arise due to a defect in membrane proteins or lipids involved in myelin compaction, P0 protein and myelin-associated glycoprotein being identified as leading candidates. Gross chemical and immunohistochemical analysis of the myelin produced in HNPP reveals no abnormalities of composition, and Schwann cell cultures from patients with HNPP showed no special features when compared to controls (Federico et al. 1989).
In 1993 a chromosome 17 deletion was found to be strongly associated with HNPP in 3 pedigrees (Chance et al. 1993), and this observation has subsequently been confirmed in both inherited and sporadic cases of HNPP (Verhalle et al. 1994; Roa et al. 1993b; Reisecker et al. 1994). Remarkably, the deleted region is seemingly identical to that normally duplicated in CMT-1A (vide supra), such that only one copy of the PMP-22 gene is present in patients with HNPP. Although attention has focused on PMP22, other genes in this deleted region or genes disrupted by the deletion could contribute to the problem. Mutation in the myelin P0 protein has recently been reported to cause a neuropathy with clinical features of CMT-1 and histological features of HNPP (Thomas et al. 1994).
19.3.4 Differential Diagnosis
Focal myelin swelling, excessive myelin infolding and outfolding, and circumferential hypermyelination are in and of themselves nonspecific, occurring to varying degrees in classic CMT-1, CMT-3, congenital dysmyelinating neuropathy, IgM paraprotein-associated neuropathy, and neuropathy with unstable myelin. Such alterations can also be present to a limited degree in patients with nonspecific neuropathy or even without neuropathy (Drac 1989). However, in the large HNPP series reviewed, above no less than 5 %, and usually a greater proportion, of internodes contained tomaculae (Drac 1989). This is beyond the frequency of such changes in patients with classic CMT-1 or Dejerine–Sottas syndrome (vide infra) or in controls (Drac 1989). No diagnostic significance can be attached to a nerve biopsy with myelin swellings or foldings on less than 1 % of internodes (Drac 1989).
A growing number of patients and pedigrees have been described where histological findings typical of tomaculous neuropathy were found in association with an atypical history: suggestive of either CMT (neuronal or hypertrophic) (Malandrini et al. 1992; Madrid and Bradley 1975; Barbieri et al. 1990; Gabreels-Festen et al. 1992b; Thomas et al. 1994) or even GBS (Joy and Oh 1989). It is unclear if these represent phenotypic variants of the same disease or if they are altogether different entities with tomaculum formation as a nonspecific response. The severity of axonal loss described in some of these reports is more typical of CMT-1 than of HNPP, and a mutation in the myelin P0 protein has been identified in one instance (Thomas et al. 1994), suggesting that at least some of these cases represent HSMN-1B rather than HNPP.
The patients designated by Gabreels-Festen and Gabreels (1993) as “CMT-1 with focally folded myelin” have myelin tomaculae that resemble those of HNPP in size, ultrastructural appearance, and frequency (vide supra). However, hypomyelination, onion bulb formation, and especially axon loss are much more prominent in this group than in HNPP. Clinically there should be little trouble distinguishing the two, as in “CMT-1 with focally folded myelin” focal palsies are not a feature, the phenotype and electrophysiological findings are more severe, and autosomal dominant inheritance is not usually seen.
In IgM paraproteinemic neuropathy, typically associated with antibody activity against myelin-associated glycoprotein, redundant myelin folds and hypermyelination very similar to that seen in HNPP are sometimes seen (Vital et al. 1989). Widely spaced, not uncompacted, myelin is present in IgM paraproteinemic neuropathy, allowing a distinction to be made purely on morphologic grounds, although clinically there is no difficulty separating the two.
Frequent “sausage-like” myelin swellings apparently typical of HNPP have been described by Said and colleagues in alcohol-associated “acrodystrophic” neuropathy (Said 1980), uremic neuropathy (Said et al. 1983), and in cytomegalovirus neuritis in a patient with AIDS (Said et al. 1991). However, the ultrastructural correlate of these swellings was not provided, other workers have not reported such data, and we have never made this observation in a number of nerves taken from patients with these diseases.
We wish to emphasize that “tomaculous neuropathy” is not synonymous with HNPP. If typical histological features of the tomaculae are present, HNPP can be diagnosed with a high degree of confidence, although a small group of patients with a chronic progressive neuropathy will remain whose classification is uncertain at present. Molecular genetics should help resolve this issue. However, detection of rare myelin tomaculae or redundant myelin loops is of little diagnostic utility.
19.3.5 Inherited Recurrent Brachial Plexus Neuropathy (Hereditary Neuralgic Amyotrophy)
Autosomal dominant inherited tendency to recurrent brachial plexopathy (inherited brachial plexus neuropathy = IBPN) has been described (Windebank 1993). Symptoms of the individual attack are identical to those of idiopathic brachial neuritis. Pain, which can be very severe, heralds the onset of paralysis by hours to days. Weakness occurs predominantly in a proximal upper limb distribution but can be present in the distal arm, cranial nerves, and lower limbs. Recovery is the rule, although this may take many months. Nosologically, this entity poses a problem, for it shares many features with HNPP. Patients with HNPP may have episodic weakness involving brachial plexus muscles, and patients with IBPN can involve the lower limbs (Dunn et al. 1978). Compression or traction on the brachial plexus may precipitate an attack in both entities. Inheritance is autosomal dominant and typical age of onset is in the second or third decade for both. Nevertheless, most neurologists separate the two based on clinical and electrophysiological criteria (Windebank 1993; Verhagen et al. 1993), although there are dissenters (Martinelli et al. 1989). Pain with paralysis is an almost invariable feature of IBPN, and preceding infection and pregnancy are common precipitating factors. Neither of these is true for HNPP. In HNPP electrophysiological tests reveal evidence of a diffuse sensorimotor polyneuropathy (Behse et al. 1972). In IBPN most studies have pointed to isolated involvement of the plexus (Windebank 1993), but not all have found this (Dunn et al. 1978).
There is little histological material on IBPN, presumably because it does not seem rational to biopsy the sural nerve in a disease of the brachial plexus. In one case studied at the Mayo clinic, no abnormality was found on exploration and fascicular biopsy of the brachial plexus (Windebank 1993). Verhagen et al. (1993) make reference to two patients in which sural nerve biopsy similarly did not reveal myelin tomaculae. Arts et al. (1983) found no myelin tomaculae in nerve biopsy and autopsy of 2 patients with IBPN. This data would suggest that IBPN is a different entity with different histological findings than HNPP.
Several pedigrees have been described with familial recurrent brachial plexopathy and myelin tomaculae on nerve biopsy (Martinelli et al. 1989; Pou Serradell et al. 1992). These patients had a painless disease, showed diffuse abnormalities on electrophysiological testing, and are thus likely to represent examples of HNPP with selective involvement of the brachial plexus. However, case 1 reported by Madrid and Bradley (1975) had prominent pain with her illness and tomaculae on nerve biopsy, as did the case we report below. The IBPN locus had been mapped to chromosome 17q24-25, and recently, genetic analysis has shown that IBPN is caused by mutation in the SEPT9 gene (Hannibal et al. 2009) which encodes a septin family member, a family of genes most involved in cytokinesis and cell cycle control. However, there remains a proportion of families with IBPN without the genetic abnormalities described above, indicating the existence of other genes involved.
Case 19.1
At the age of 12, this patient developed weakness and pain involving the right shoulder. He was unable to lift the arm due to weakness, and some muscle atrophy was subsequently noted. A diagnosis of polio was made. The weakness gradually improved over several months. A right wrist drop developed at the age of 17 and recovered over 6–12 months. The patient was fit enough to join the army subsequently. Intermittent paraesthesias, particularly involving the ulnar nerve territory and more prominent on awakening, plagued the patient over during his third decade. A few moments of pressure on any nerve resulted in numbness and tingling lasting for minutes to hours. After crossing his legs, he often found that he experienced difficulty walking for a few minutes. At the age of 38, the patient experienced sudden onset of pain and weakness in the left shoulder girdle, and a diagnosis of brachial neuritis was ultimately made. Despite initially complete paralysis of arm abduction and definite atrophy, full recovery was eventually attained. At the age of 43 the patient presented for neurological consultation with a complaint of bilateral symmetrical hand and foot weakness and clumsiness.
The patient had no siblings but two sons were said to be normal. His father was said to have “numb hands” and wasting of small hand muscles. Neurological examination documented normal muscle bulk, normal power, normal reflexes, and a slight multimodality decrease of sensation in a right ulnar nerve distribution.
Nerve conduction studies showed focal conduction slowing across the right ulnar nerve at the elbow, the right median nerve at the wrist, and the right peroneal nerve across the fibula. All F-wave responses were slowed. Motor conduction velocities of the ulnar and median nerves in the forearms were on the lower limit of normal, and the peroneal nerve below the fibula was definitely slowed at 33 m/s. Sensory potentials were moderately delayed and diminished in amplitude. Subsequent examination of the patient’s sons showed no clinical abnormalities, but conduction slowing was found across some sites of compression.
Biopsy confirmed a clinical diagnosis of HNPP. Subsequently the patient underwent bilateral carpal tunnel release and transposition of the right ulnar nerve, resulting in considerable improvement in his hand symptomatology and function (clinical material courtesy of Dr. P. Ashby).
19.4 CMT-2
19.4.1 Clinical Manifestations
Neuronal (type 2) CMT (CMT-2) is the typically autosomal dominant (50–70 %) neuronal form of the disease showing motor nerve conduction velocities of 38 m/s or greater, with 25–50 % sporadic and 5 % recessive cases in the largest series (Harding and Thomas 1980c; Bouche et al. 1983; Berciano and Combarros 1990). Segregation analysis suggests that 25 % of sporadic cases are recessive and the rest new dominant mutations (Harding and Thomas 1980c). Genetic analysis of autosomal dominant CMT-2 now has identified 19 gene defects and 5 abnormal genes in autosomal recessive CMT-2. Its prevalence is about one-third that of CMT-1 and CMT-2A; its most frequent form (20 % of CMT-2 cases) is associated with a missense mutation of Mfn2 (mitochondrial fusion protein 2 gene). Mitofusin (Mfn2) is a mitochondrial membrane protein that participates in mitochondrial fusion in mammalian cells and in pyruvate, glucose, and fatty acid oxidation, defects of which culminate in reduced mitochondrial membrane potential. Experimental data support the notion that Mfn2 loss of function causes axonopathy by impairing energy production along the axon which may reflect abnormal tethering of mitochondria to ER (Pich et al. 2005). Type 2A CMT is clinically similar to CMT-1, although on average the phenotype is milder (peripheral nerves are not enlarged) and the disease develops later (mean age of onset 20 years), there is less prominent involvement of upper limbs (particularly the hands) and less ataxia, tendon areflexia, and sensory loss. Mutations in the RAB7 (small GTPase late endosomal protein) gene cause CMT-2B, a predominantly sensory neuropathy complicated by chronic foot ulcers and amputations (Lawson et al. 2005). Type CMT-2D is associated with mutations of the GARS (glycil tRNA synthetase) gene and consists of upper extremity wasting and weakness, with corresponding sensory impairment; lower extremities are involved to a lesser degree (Lawson et al. 2005). Mutations in the NEFL gene are associated with axonal neuropathy and designated as CMT-2E. This condition is characterized by variable severity and sensory ataxia. NCS may show preservation of normal values or slight slowing. Pathological findings are of interest in that they reveal the presence of giant axons with attenuated myelin and containing aggregates of disorganized neurofilaments (Pareyson et al. 2013).
Autosomal recessive forms of CMT-2 are called autosomal recessive CMT (AR-CMT). Recessive mutations of the LMNA (laminin A/C) gene are the cause of the axonal neuropathy in AR-CMT-2 (formerly known as CMT-4C and CMT-2B1). Whether a neuronal X-linked form exists is unclear (Hahn 1993; Dyck et al. 1993). Clinical manifestations are much like those of CMT-1, summarized above. No clinical features definitively distinguish the two, although onset in the first decade and palpable nerves suggest the hypertrophic form, and variations in distribution of atrophy, weakness, and reflex changes may be helpful (Hahn 1993). As with CMT-1, examination of “normal” family members is essential because the disease may be asymptomatic.
Patients with childhood onset are more severely affected, often resulting in debilitation by adulthood (Ouvrier et al. 1981; Gabreels-Festen et al. 1991). Most of these are probably genetically distinct from dominantly inherited CMT-2. From a total of 29 cases with childhood onset and a severe clinical course (Gabreels-Festen et al. 1991; Ouvrier et al. 1981), only 3 had a dominant inheritance pattern, 8 had similarly affected siblings and normal parents suggesting recessive inheritance, and most were sporadic.
Nerve conduction studies are usually the basis for a definitive distinction of CMT-2 from CMT-1, but rarely there are situations where the conduction velocities are slowed, presumably due to the selective and sometimes very severe loss of large (rapidly conducting) myelinated fibers (vide infra). Conduction velocity is usually normal or near normal, with reduced amplitudes suggesting loss of axons. EMG shows changes of very chronic denervation. CMT-2 has been reviewed by Hahn (1993), Dyck et al. (1993), and genetic analysis (Rossor et al. 2013; Saporta and Shy 2013; Sagnelli et al. 2013; Tazir et al. 2013; Vallat et al. 2013).
19.4.2 Pathology
There are several large CMT-2 biopsy series (Behse and Buchthal 1977; Gherardi et al. 1983; Ouvrier et al. 1981; Berciano et al. 1986; Gabreels-Festen et al. 1991).
19.4.2.1 Light Microscopy
Histologically, this disorder displays some neuron loss in ventral horn and spinal ganglia of the lumbosacral region and tract degeneration in the gracilis fasciculi. In peripheral nerves there is mostly chronic axonal loss affecting large myelinated fibers, axonal atrophy as evidenced by an increase in the axon caliber/myelin thickness ratio, abundant regenerating clusters, and no hypertrophy. Endoneurial edema and fascicular enlargement are not features of CMT-2 biopsies, but atrophy of the nerve fascicle is occasionally seen. In longstanding cases fibrosis is prominent. Myelinated fiber numbers are usually less dramatically reduced than in CMT-1 of comparable duration, with a more selective loss of large MFs. Unmyelinated fibers are spared or only slightly reduced in number with autosomal dominant adult cases, but may be significantly reduced in recessive/sporadic childhood cases. Interpretation of axon numbers is difficult due to the high frequency of regenerating clusters. In the recessive and infantile onset forms, MF loss can be very severe, often with no axons greater than 6 μm in diameter remaining (Gabreels-Festen et al. 1991). In one extreme case all remaining MFs were within the 0.6–1.8 μm diameter size range! Despite the severity of axonal loss, Wallerian degeneration is a rarity, even in the more severe recessive/childhood cases.
In contrast to CMT-1, regenerating clusters are quite frequent in dominant CMT-2, quantified at 20–400/mm2 (normal being <20/mm2) (Gherardi et al. 1983; Behse and Buchthal 1977) (Fig. 19.11a–d, arrows, d). This has also been expressed as the “cluster ratio,” measuring the number of regenerating clusters per 1,000 MFs, and values in dominant CMT-2 range from 2.4 to 38 (Ouvrier et al. 1981; Gherardi et al. 1983; Gabreels-Festen et al. 1991), with normal being less than 2, often 0, in adults (extrapolated from data of Gherardi et al. (1983) Behse and Buchthal (1977)). In the severe recessive or sporadic childhood cases described by Gabreels-Festen et al. (1991) and Ouvrier et al. (1981), regenerating clusters were usually not seen. One exception was an autosomal dominant case (Gabreels-Festen et al. 1991), which had a cluster ratio of 8.1. An interesting observation is that cluster numbers diminish more distally in a nerve, and with increased disease duration, presumably because the distal axon is “sicker” and less able to regenerate (Berciano et al. 1986; Gherardi et al. 1983).
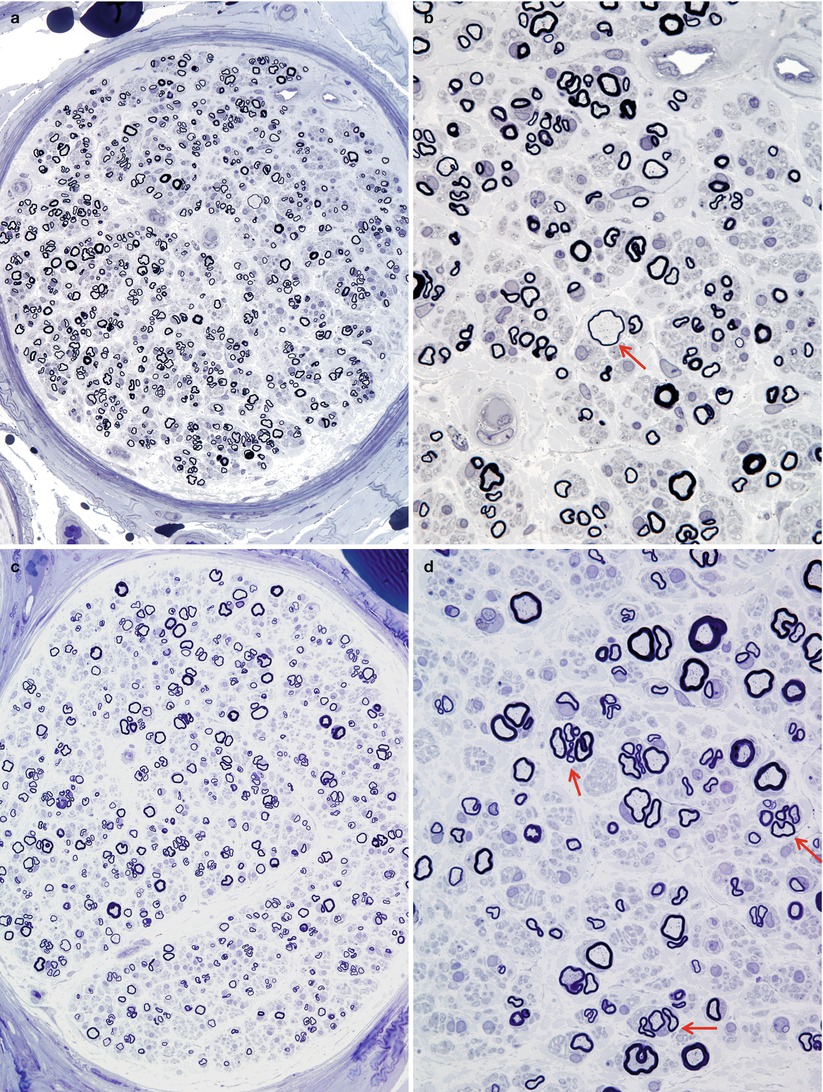
Fig. 19.11
Two cases of hereditary axonal neuropathy show moderate reduction in number of myelinated fibers and regenerating clusters. Rare thinly myelinated swollen axons are a nonspecific finding (arrow, b). Biopsy from a 28-year-old woman with dominantly inherited CMT-2 of 18 years duration is depicted in figs (a, b). Biopsy from a 58-year-old woman with dominantly inherited axonal neuropathy and spastic paraparesis, likely CMT-5, is illustrated in figs (c, d). Note the regenerative clusters (arrows, d) (1 μm thick plastic section. Magnification: a, c 400×; b, d 1,000×)
The presence of MFs with inappropriately thin myelin sheaths is variable, but can be surprisingly prominent for a neuropathy which is not regarded as having a demyelinating component. Fiber teasing may demonstrate an increase in segmental myelin alterations above normals, and it is presumed that these are secondary to axonal atrophy (Berciano et al. 1986; Dyck et al. 1993).
19.4.2.2 Electron Microscopy
Well-formed onion bulbs are absent or rare in CMT-2, even by ultrastructural criteria. Those cases where their presence has been noted may represent secondary demyelination or “pseudo”-onion bulbs (Tome et al. 1979; Hahn 1993). The latter explanation also probably accounts for the presence of inappropriately thin myelin sheaths. Nonspecific axonal alterations may be seen in myelinated and unmyelinated fibers (Vital et al. 1979; Julien et al. 1988; Yasuda et al. 1990; Hahn et al. 1990), but are not a dominant feature.
One pedigree with neurological features typical of CMT-2, some patients having evidence of cardiomyopathy, has been reported to show giant axonal swellings due to massive focal accumulations of neurofilaments, similar to that seen in hexacarbon toxicity (Chap. 18) and recessive/sporadic giant axonal neuropathy (Vogel et al. 1985). The X-linked family described by Hahn et al. (1990) had features typical of CMT-2, supporting the existence of X-linked neuronal CMT-2.
19.4.3 Pathogenesis
The genetics of the various forms of CMT-2 (Rossor et al. 2013) suggest a variety of pathogenetic mechanisms ranging from altered mitochondria and mitochondria-based metabolism of axons/Schwann cells to axonal cytoskeleton, endosomal functions, and others. Autopsy and teased fiber studies support the hypothesis that a distal axonopathy with axonal atrophy is the fundamental pathogenic mechanism (Berciano et al. 1986; Yasuda et al. 1990; Dyck et al. 1993).
19.4.4 Differential Diagnosis
Individually, the histological findings of CMT-2 are nonspecific. However, a biopsy showing diffuse loss of myelinated fibers, with relatively selective large fiber involvement, no active degeneration, and very prominent regenerating cluster formation, is highly suggestive of the diagnosis. In theory, a similar picture may be seen in other very longstanding axonal neuropathies such as diabetes, chronic renal failure, or long duration toxic exposures, but in our experience these always show a degree of active (Wallerian) degeneration that is out of keeping with the diagnosis of CMT-2.
19.5 Autosomal Dominant Intermediate CMT
Validity of the classification of CMT into neuronal and hypertrophic types is strongly supported by the bimodal nerve conduction velocity distribution (Dyck et al. 1993; Buchthal and Behse 1977). As discussed above, this usually correlates well with histological findings: onion bulbs and no regenerating clusters in CMT-1 and the reverse in CMT-2. However, Madrid, Davis, and Bradley (1977; Bradley et al. 1977) found clinical, electrophysiological, and histological evidence for an “intermediate group” of CMT patients. The most distinctive features were motor conduction velocities in the 25–45 m/s range, biopsies revealing a combination of onion bulb and regenerating cluster formations, and ultrastructural evidence of active axonal degeneration. Other authors have found that nerve conduction velocities (Brust et al. 1978) and histological observations (Rossi et al. 1985; Gherardi et al. 1983) do not invariably support easy division into neuronal and hypertrophic forms. We have occasionally examined biopsies in patients with genetically determined neuropathy showing both OBs and denervated bands (Figs. 19.12 and 19.13). However, the biopsies reported as “intermediate” by Madrid and colleagues (1977) were said to show “not infrequent” active axonal degeneration and axonal ultrastructural abnormalities. This is a finding that we have never observed in patients with any type of CMT and which we would not consider compatible with the diagnosis, unless an additional disease was superimposed. None of the other large nerve biopsy series in CMT-1 or CMT-2 report significant active axonal degeneration, even in severe infantile and childhood-onset neuronal cases (Ouvrier et al. 1981; Gabreels-Festen et al. 1991).
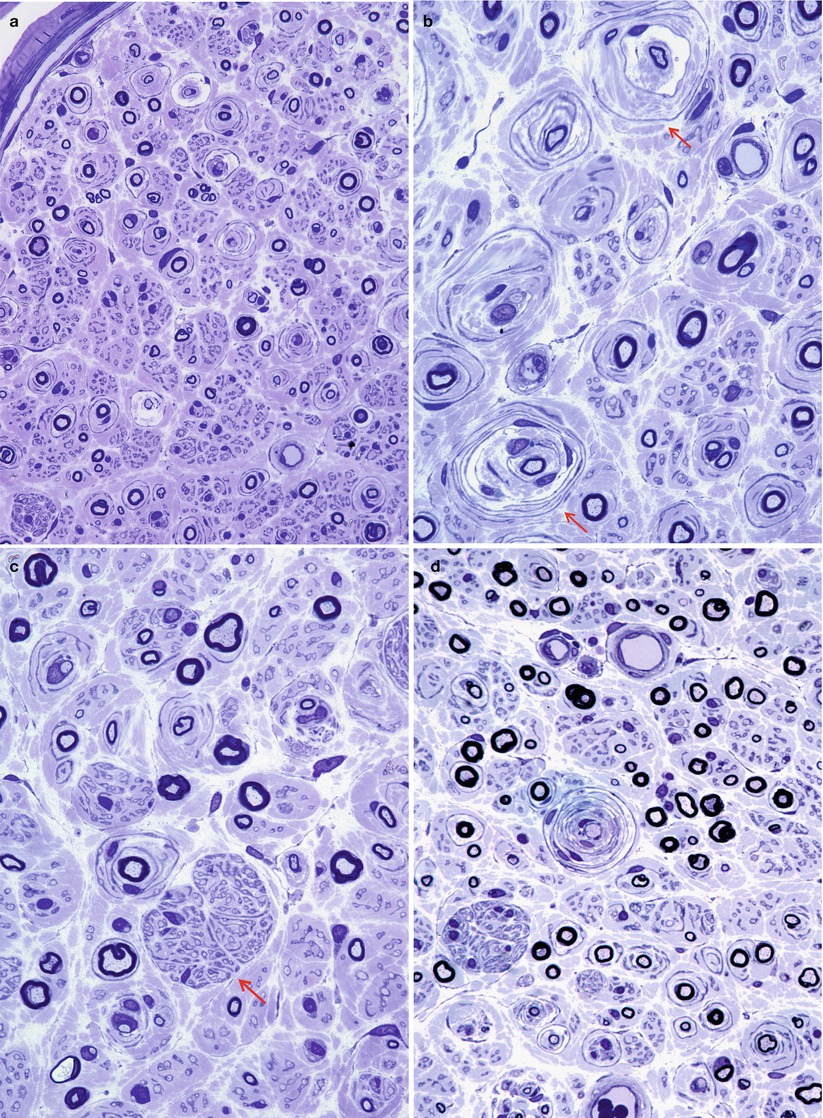
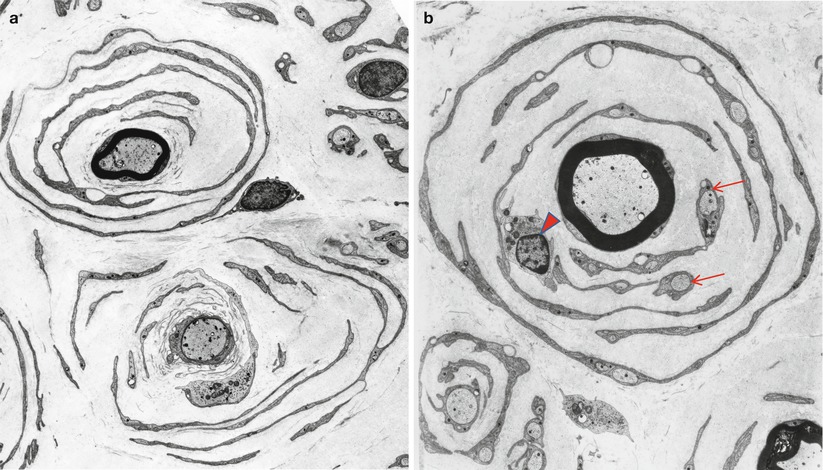
Fig. 19.12
CMT-“intermediate”: a 30-year-old woman with familial neuropathy and mildly reduced conduction velocity, inheritance uncertain. Nonuniform pattern of pathological alterations is shown (a). While some fibers are normal, others demonstrate either typical or giant onion bulbs (arrows, b) or large denervated band (arrow, c) or both (d) (1 μm thick plastic section: a, 400×; b–d, 1,000×)
Fig. 19.13
CMT-“intermediate”: variation in morphology of onion bulbs. Central axons are surrounded by short segments of redundant basal lamina, one denuded and one thinly myelinated (a). In (b) the central fiber is fully myelinated and miniature axons can be seen enclosed within Schwann cells of the onion-bulb rings (arrows). The significance of a lymphocyte (arrowhead) within the OB is undetermined – inflammation was absent otherwise. Same case as Fig. 19.12 (Magnification: a, 3,360×; b, 4,080×)
Genetic analysis of such patients has identified dominant gene defects (DNM2, YARS, MPZ, INF2, GNB4) and recessive (GDAP1, KARS, PLEKHG5) forms, different gene defects involving a wide variety of targeted genes (Rossor et al. 2013).
An excellent correlation exists between electrophysiological and histological subdivision of autosomal dominant CMT in types 1 and 2 (Buchthal and Behse 1977), but there are rare exceptions (Harriman and Currie 1979; Gherardi et al. 1983). A biopsy showing prominent onion bulbs may come from a patient with conduction velocities greater than 38 m/s, and conversely a patient with markedly showed conduction may show the histological picture of neuronal CMT. The latter may be ascribed to a severe loss of axons, especially large MFs, resulting in a surviving population that conducts slowly. Some of the former might fall under the “intermediate” group or may represent a disjunction between the pathology of motor and sensory nerves.
19.6 CMT-Associated Demyelinating Neuropathies of Early Onset
The majority of perinatal early-onset hereditary neuropathies represent demyelinating phenotypes (58 %) with slowed motor nerve conduction velocities, indicative of demyelination or amyelination (Baets et al. 2011). Early-onset hereditary polyneuropathies are not a single entity but rather represent a broad and heterogeneous clinical and genetic spectrum of disorders, which include both dominant and recessive mutations in multiple genes (Baets et al. 2011). Several factors militate against the formulation of a heuristic classification of congenital neuropathies; these include as follows: a single gene abnormality can cause multiple phenotypes of variable severity, a single phenotype can be caused by mutations in different genes, the same phenotype may result from a de novo dominant mutation or from a recessive mutation, and that many of the children with early-onset congenital neuropathy remain without a specific diagnosis after multiple testing. (Yiu and Ryan 2012; Baets et al. 2011). For current classifications of demyelinating prenatal and infantile developmental neuropathies, the reader is directed to Yiu and Ryan (2012), Landrieu and Baets (2013), and Baets et al. (2011).
Congenital hypomyelinating neuropathy (CHN), Dejerine–Sottas Disease (DSD), also termed CMT-3, and CMT-4 (also designated as autosomal recessive CMT-1) are forms of demyelinating CMT with prenatal or infantile development. Most infants with early-onset neuropathies may display one of two phenotypes. The first group is genuinely congenital and is characterized by perinatal onset with hypotonia, muscle weakness at birth, arthrogryposis, respiratory insufficiency, and marked conduction slowing to 6–12 m/s or less (Ouvrier et al. 1987; Benstead et al. 1990). These patients experience delayed development. The second and most common group, after an uneventful neonatal period, manifests in infancy with motor delay and foot deformities and show slow progression into adult life (Yiu and Ryan 2012).
Dejerine–Sottas syndrome, formerly designated CMT-III (CMT-3 and DSD), is the first and most clearly defined subgroup of CDN; however, it shows substantial clinical and pathological overlap with CHN. Molecular genetics has shown that both are in fact at the severe end of the CMT-1 disease spectrum (Landrieu and Baets 2013). Noteworthy is the fact that of the original two siblings reported by Dejerine and Sottas, one showed disease onset in infancy and the other at the age of 14 years (Dyck et al. 1993).
Dejerine–Sottas and CHN are genetically complex syndromes (Gabreels-Festen 2002) that have been reported as sporadic or autosomal recessive. They are associated with de novo dominant mutations involving at least 3 different genes (EGR2, PMP22, MPZ) and recessive mutations in EGF2 and periaxin genes. In its classic form, CMT-3 is a severe hypertrophic polyneuropathy of early onset (first 2 years of life) with delayed milestones, progressive and debilitating clinical course, with half of patients becoming wheelchair bound by the age of 10 years (Dyck et al. 1993; Anderson et al. 1973; Ouvrier et al. 1987; Guzzetta et al. 1982; Weller 1967). Nerves are palpable in most patients, CSF protein is elevated, and motor nerve conduction velocity is below 6–12 m/s. There is clinical and electrophysiological overlap between DSS and CMT-1 given that they share mutations of the same group of genes. Although debility in DSD is more severe than for CMT, DSD is now placed by many among the CMT-1 group.
19.6.1 Light Microscopy
Nerve biopsy shows large numbers of onion bulbs, endoneurial fibrosis, loss of axons, and, specifically, hypomyelination: Many fibers that, on the basis of axon diameter, should be myelinated lack myelin completely or show disproportionately thin myelin sheaths (Fig. 19.14a, b) (Guzzetta et al. 1982). Histological features are reminiscent of dominant CMT-1, but more severe as regards nerve enlargement, onion bulb formation, and axonal loss (Fig. 19.14). As many as 50 % of internodes may be demyelinated on cross sections and even more dramatically on teased fibers (Dyck and Gomez 1968; Dyck et al. 1970, 1971b; Ouvrier et al. 1987). Despite the large number of denuded axons, macrophages containing myelin debris are not prominent. The most salient observation, however, is that when axons do have a myelin sheath, it is always inordinately thin, regardless of axon size (Figs. 19.14 and 19.15). The myelinated axon population is reduced, sometimes very severely, with a marked shift towards small diameters. There can be overlap between the degree of axon loss seen in autosomal dominant CMT-1 and DSS in patients of roughly the same age (Ouvrier et al. 1987). Unmyelinated axon counts seem normal, but with the shift of the myelinated axon population towards small fiber size and the florid demyelination, it may be difficult to classify small nonmyelinated axons.
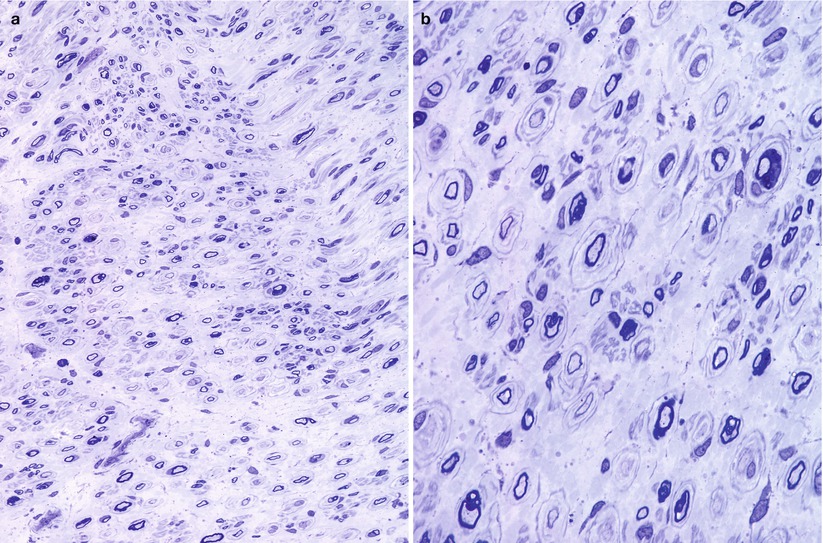
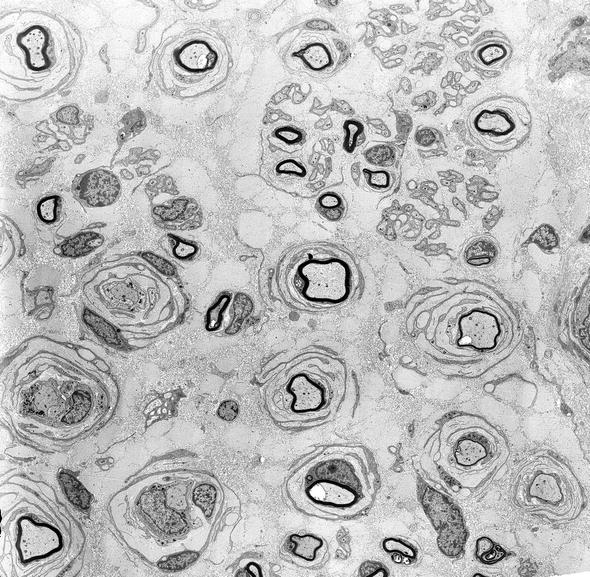
Fig. 19.14
CDN-Classic Dejerine–Sottas (CMT-3): note that none of the fibers are fully myelinated and many are amyelinic, yet no lipid debris is seen. Onion bulbs with thin leaves are prominent (b) (1 μm thick plastic section, magnification: a, 200×; b, 1,000×) (Tissue courtesy of Dr. V. Jay, Toronto)
Fig. 19.15
CDN-Classic Dejerine–Sottas: low-power view showing lack of full myelination and many onion bulbs containing demyelinating and remyelinating fibers (1,800×)
19.6.2 Electron Microscopy
Onion bulbs are a prominent feature and may reach large proportions. The lamellae are made up of concentric imbricated layers of Schwann cell processes, as well as redundant basement membranes (Figs. 19.15 and 19.16a). Hypomyelination is the rule, with only 10–20 myelin lamellae per myelinated axon, regardless of its diameter (Dyck et al. 1971a). Ouvrier et al. (1987) reported that this represents the most consistent means of distinguishing morphologically between autosomal dominant CMT-1 and CMT-3: The g-ratio (see Chap. 3) was never above 0.69 in 10 cases of childhood-onset CMT-1 and never below 0.81 for 6 cases of Dejerine–Sottas. Myelin debris may be seen within Schwann cells. A slight increase in collagen pockets and denervated Schwann cell bands, subtle indicators of mild unmyelinated axon loss, has been reported (Ouvrier et al. 1987).
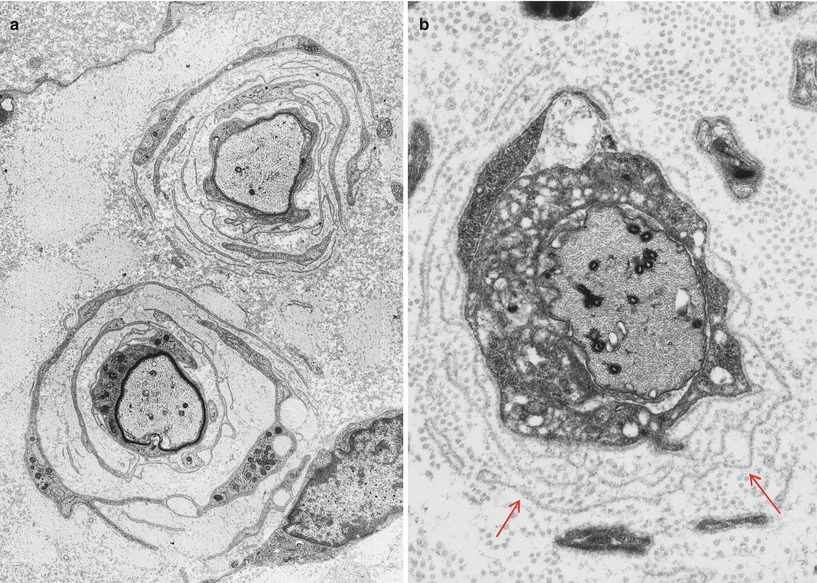
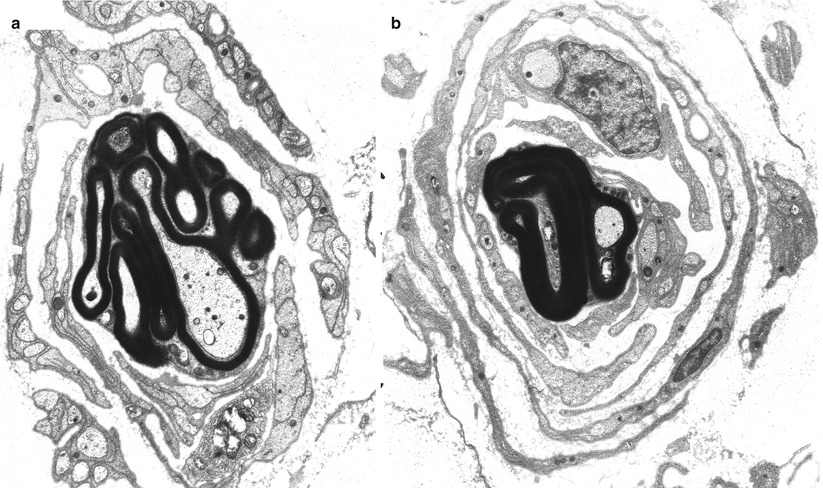
Fig. 19.16
CDN-Classic Dejerine–Sottas: (a, b) basal lamina rings form a prominent part of these onion bulbs (Magnification a, 7,500×; b, 10,000×)
Fig. 19.17
CDN-Focally folded myelin: (a, b) two onion bulbs are centered by axons that display elaborate outer redundant loops of myelin (Magnification a, b, 7,500)
19.6.3 Pathogenesis
Since PMP duplications and point mutations are also associated with CMT-1A (vide supra), it is evident that the clinical and histological phenotype is dependent on additional factors, perhaps the site of mutation in the gene. Mutations in myelin P0 protein have been found in patients satisfying clinical or histological criteria for DSS (Hayasaka et al. 1993d; Himoro et al. 1993). One of the three mutations occurred in the transmembrane domain of the P0 protein and the others in the extracellular variable immunoglobulin-like region. In one instance these workers showed that P0 was expressed in normal quantities in peripheral myelin (Tachi et al. 1994). As with the PMP-22 mutations, mutations in P0 are also associated with the CMT-1 phenotype (vide supra: CMT-1B).
19.6.4 CMT Type III with Basal Lamina Onion Bulbs (CMT-3 BLOB)
Of historical interest was the proposed grouping, by Gabreels-Festen and Gabreels (1993), of patients under the description of CMT type III with basal lamina onion bulbs (CMT-3-BLOB). Their review of the literature identified about 30 such cases (Harati and Butler 1985; Joosten et al. 1974; Lutschg et al. 1985; Balestrini et al. 1991; Ono et al. 1982; Moss et al. 1979; Vital et al. 1987; Vallat et al. 1987; Lyon 1969; Boylan et al. 1992; Guzzetta et al. 1982; Kennedy et al. 1977). Onset is most often at birth or infancy, with a variable disease severity ranging from arthrogryposis to survival to adult life with moderate disability. Conduction velocity is often below 6 m/s. Thus, these patients are clinically indistinguishable from the classic DSS syndrome discussed above.
19.6.4.1 Pathology
Light microscopic examination may show enlarged nerve fascicles, with an increase in endoneurial matrix (Joosten et al. 1974). Only a few thinly myelinated fibers remain, but the axonal loss is not as severe as a quick glance would indicate, for the majority of axons in the nerve fascicle are demyelinated. In the various case reports, axon numbers ranged from severely reduced (Gabreels-Festen and Gabreels 1993) to relatively preserved (Harati and Butler 1985; Boylan et al. 1992). Large axons seem to be more severely affected.
Ultrastructural examination reveals attenuated myelin, which occasionally may be uncompacted. The characteristic finding is that many axons, regardless of whether they are myelinated or not, are surrounded by basal lamina rings (arrow, Fig. 19.16b), with few or no classic onion bulb structures. Very thin Schwann cell processes may be found at the periphery. Sometimes the Schwann cell does not fully surround the naked axon, and the axon is adjacent to basement membrane without intervening Schwann cell cytoplasm. The rare regenerating cluster may be seen, as well as infrequent redundant myelin folds. Myelin debris and active demyelination in the presence of an intact axon has been described in some of these cases (Joosten et al. 1974; Balestrini et al. 1991; Moss et al. 1979; Lyon 1969; Kennedy et al. 1977).
19.6.4.2 Discussion
CMT-3-BLOB and classic DSS share many features including typical age of onset, variation in severity of disease, similar inheritance patterns, very slow conduction velocities, frequent demyelination with myelin debris, and striking hypomyelination. The main distinction appears to be the presence of numerous basement membrane onion bulbs. However, these are also seen in “classic” DSS (Dyck and Gomez 1968; Ouvrier et al. 1987), and the difference seems to be only a matter of degree. BLOBs are also part of the CMT-4A and CMT-4C groups, which have GDAP1 (ganglioside-induced differentiation-associated protein 1) and SH3TC2 gene defects identified. Many workers (Dyck et al. 1993; Ouvrier et al. 1987) do not identify two distinct groups of patients with Dejerine–Sottas syndrome.
19.6.5 CMT-1 with Basal Lamina Onion Bulbs
Gabreels-Festen and colleagues suggested the delineation of a group of patients in which BLOBs are prominent, but which are distinguished histologically from the two groups previously mentioned by virtue of milder myelin abnormalities (Gabreels-Festen et al. 1992a; Gabreels-Festen and Gabreels 1993). These authors identified several similar cases in the literature (Meier et al. 1976; Nordborg et al. 1984; Smith et al. 1980). Clinically the patients suffer from a disease intermediate in severity between typical Dejerine–Sottas syndrome and CMT-1. Conduction velocities are usually in the 10–30 m/s range. All reported cases have been sporadic or autosomal recessive. Testing for the CMT-1A mutation has been negative (Gabreels-Festen and Gabreels 1993), but there may be overlap with CMT-4A and CMT-4C groups which have GDAP1 (ganglioside-induced differentiation-associated protein 1) and SH3TC2 gene defects identified.
Biopsy may show an increased endoneurial area with interstitial edema. There is reduction in myelinated fiber density, but unmyelinated fibers are relatively spared. Classic onion bulb formations are small and less frequent than BLOBs. Occasional myelin tomaculae are present. Teased fibers show segmental myelin changes without signs of active axonal degeneration. The major distinguishing point from the Dejerine–Sottas picture is that far fewer demyelinated axons are seen, and there is no evidence of the severe hypomyelination that characterized DSS. When measured, the g-ratio has been in the range 0.64–0.77.
The patients reported by Nordborg et al. (1984) showed intra-axonal lamellated inclusions in unmyelinated fibers and less often in myelinated fibers, as well as frequent redundant myelin loops.
The existence of yet another morphologically unusual variant of CDN, designated “autosomal recessive CMT with focally folded myelin (FFM)” was postulated by Gabreels-Festen and colleagues (Gabreels-Festen et al. 1990), who identified similar cases from the literature (Barbieri et al. 1994; Lutschg et al. 1985; Routon et al. 1991; Vital et al. 1987; Ouvrier et al. 1990 case 14.3; Nordborg et al. 1984). Umehara et al. 1993 described a dominantly inherited case of motor and sensory neuropathy with excessive myelin folding complex. Symptoms are present at birth or appear within the first year, with slow progression. Some are wheelchair bound in childhood, while others retain the ability to walk well into adulthood. Conduction velocities have been above and below the cutoff of 10–12 m/s usually used to delineated Dejerine–Sottas syndrome. CSF protein is usually normal.
19.6.5.1 Pathology
Microscopic examination reveals two populations of axons. Many are thinly myelinated or amyelinic, and the g-ratio is frequently in the range associated with DSS. A second group of axons demonstrate folded redundant myelin loops or circumferential hypermyelination with occasional signs of degeneration. Onion bulb formations are present, in numbers seemingly proportional to the patient’s age. In patients reaching adult life, onion bulbs are more frequent, may attain a substantial size, and are often denervated. Classic OBs and BLOBs can be seen. The myelinated axon population is normal in very young patients and declines with age, with large myelinated axons being more severely depleted. Myelin debris may be seen. Unmyelinated axons are usually unscathed. Teased fiber studies show that myelin “tomaculae” are present in many as 50–75 % of internodes.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


