CHAPTER 200 Ependymoma
Clinical Overview
Roughly 10% of pediatric brain tumors are classified as ependymoma, and the mean age at diagnosis is 4 to 6 years.1–5 About 5% of patients with ependymoma present with evidence of leptomeningeal dissemination at the time of diagnosis, as determined by a combination of magnetic resonance imaging (MRI) with contrast enhancement and cerebrospinal fluid (CSF) sampling.6 Historical 5-year survival estimates range from 50% to 64%; progression-free survival estimates are lower at 23% to 45%.2,3,7–9 Tumor typically recurs at the site of the original tumor, but in roughly 20% of cases there is isolated metastatic recurrence. The median time to recurrence is 13 to 25 months, and most children who are disease free at 5 years are cured (although late recurrences have been described).2,3,7–9
In childhood, 90% of ependymomas are located intracranially: one third supratentorially, and two thirds infratentorially.10 Sixty percent of supratentorial ependymomas arise contiguous with or adjacent to the ventricular system (lateral or third ventricle); the other 40% arise in areas remote to the ventricles and their ependymal surface (Fig. 200-1). Infratentorial (posterior fossa) ependymomas arise from the floor (60%), lateral aspect (30%), or roof (10%) of the fourth ventricle (Fig. 200-2).11,12
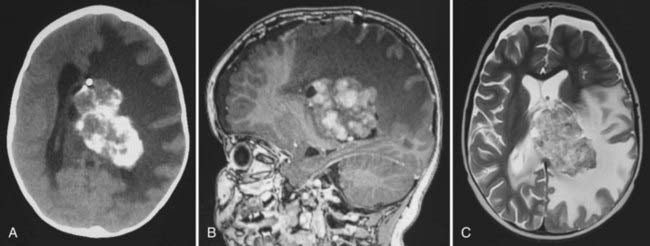
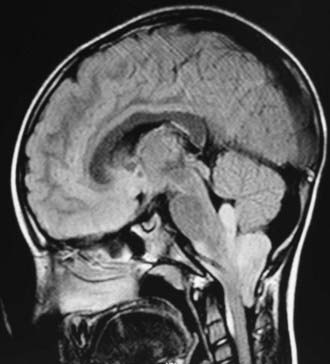
Prognostic Factors
Extent of Resection
The single most important determinant of outcome in pediatric ependymoma is the extent of surgical resection, emphasizing the role of the pediatric neurosurgeon. The 5-year survival rate in children who receive gross total resection is 67% to 80%, and the 5-year progression-free survival rate is 51% to 75%.2–5,7–9,13–21 In striking comparison, the 5-year survival rate of children who undergo subtotal resection ranges from 22% to 47%, with a 5-year progression-free survival ranging from 0% to 26%. Many authors have advocated repeated resection when residual tumor is found on postoperative imaging.12,15,22
MRI with contrast is recommended within 72 hours of resection to avoid confusing postoperative artifacts. Ependymoma resections are classified as gross total, near total, subtotal, and biopsy. Gross total resection is defined as the absence of tumor on postoperative MRI, even if microscopic disease (visualized with the operating microscope during surgery) is still present. Near total resection is arbitrarily defined as less than 1.5 cm3 residual tumor on postoperative imaging (similar to the values quoted for medulloblastoma) or 0.5 cm residual thickness of tumor bed enhancement.9 Subtotal resection is defined as remaining gross residual tumor that is visible to the surgeon during surgery or on postoperative neuroimaging. Because minimal residual disease can be cured with radiation, it is not necessary to perform extensive, aggressive resections that put the patient at risk.
Histologic Grade
The value of histologic grading in determining the prognosis of ependymoma patients is controversial.4,9,16,18,19,23–26 Compared with classic ependymoma, anaplastic ependymoma exhibits histologic evidence of advanced anaplasia, including nuclear atypia, marked mitotic activity, high cellularity, and microvascular proliferation or necrosis. Analysis of a group of 50 contemporary ependymoma patients from St. Jude Children’s Research Hospital showed that tumor grade was significantly related to progression-free survival after irradiation (P < .001).15 In this group of patients, the 2-year event-free survival estimate was 32% ± 14% for children with anaplastic ependymoma, compared with 84% ± 7% for children with classic ependymoma. This association remained significant after adjusting for age younger than 3 years, chemotherapy, and extent of resection. Of note, supratentorial ependymoma is more likely to show anaplastic histology than is posterior fossa ependymoma.
Age at Diagnosis
Children younger than 3 years at the time of ependymoma diagnosis have a poorer prognosis than older children. Younger children are less resilient and more likely to suffer complications from surgery, radiation, and chemotherapy. As a rule, radiation is not given to children younger than 3, or it is given in lower doses supplemented by chemotherapy. A study by the Pediatric Oncology Group showed a 63% 5-year survival for children aged 24 to 35 months (radiation delayed 1 year), but only a 26% 5-year survival for children aged 0 to 23 months (radiation delayed 2 years).27 These data emphasize the importance of radiation treatment for increasing the survival rates in children and that the poor prognosis in very young children relates at least in part to our inability or unwillingness to irradiate the developing brain.28,29 Although it has been suggested that higher doses of radiotherapy may improve the outcome for ependymoma patients, this has not been shown in a randomized trial.30
Pathology
In the World Health Organization (WHO) classification of brain tumors, a grade II ependymoma can be diagnosed only when mitoses are rare or absent; occasional foci of palisading necrosis are allowed, as are nodules with increased cellularity and mitotic activity.31 Anaplastic ependymoma is a grade III lesion in the WHO classification.32 It is diagnosed in tumors with increased cellularity, brisk mitotic activity, vascular proliferation, pseudopalisading necrosis, clear ependymal differentiation, perivascular pseudorosettes, cytologic atypia, and microvascular atypia.31 Areas of hypercellularity may be diffuse or focal and may form well-circumscribed regions abutting those of lower cellularity. Additionally, areas of cytologic atypia, including increased nuclear-to-cytoplasmic ratios and cellular pleomorphism, may be seen. Anaplastic regions often have a higher mitotic rate, although no specific threshold for a diagnosis of anaplastic ependymoma is widely accepted. Neither focal areas of atypia nor brisk mitotic activity is sufficient to make a diagnosis of anaplastic ependymoma. It is unclear whether anaplastic ependymoma arises from the progression or malignant degeneration of classic ependymoma or whether it occurs de novo.
Histologic grading and its clinical significance are difficult to assess, and agreement among pathologists when assigning a tumor grade to ependymoma is poor. Failure to find true ependymal rosettes or perivascular pseudorosettes is associated with a poor prognosis in children with ependymoma.13 Two separate groups have reported that the combination of necrosis, endothelial proliferation, and a mitotic index greater than 5 is a negative predictive factor for both overall and progression-free survival.13,33 Others have shown an inverse correlation between mitotic rate and survival in ependymoma.34 Ki-67 and MIB-1 immunolabeling methods to measure mitotic activity have also been used to show an inverse correlation between mitotic rate and outcome in two series of pediatric ependymoma.13,21,33,35 Another group of investigators found that mitotic rate was important only in determining the prognosis of supratentorial ependymoma.36
Hereditary Tumor Syndromes
The vast majority of ependymomas occur sporadically, although some well-documented associations with hereditary tumor syndromes have been described. Intramedullary spinal ependymoma is well known to occur in patients with neurofibromatosis type 2 (NF2) (Fig. 200-3). In contrast, patients with NF2 are probably not at increased risk of developing intracranial ependymomas. The same biology is found in patients with sporadic (nonfamilial) intramedullary ependymomas who have somatic mutations of the NF2 gene. Mutations of the NF2 gene are not found in intracranial ependymomas, myxopapillary ependymomas, or tanycytic ependymomas.37,38 The NF2 gene is located on chromosome 22q. Although intracranial ependymomas are well known to show a loss of genetic material on 22q, they do not harbor NF2 mutations, suggesting the existence of another distinct ependymoma tumor suppressor gene in this region of the genome.
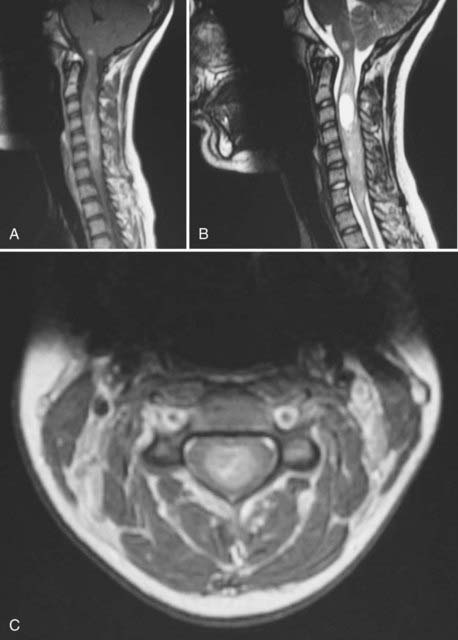
Ependymoma has also been reported in patients with the Li-Fraumeni familial cancer syndrome (due to a germline mutation in the p53 tumor suppressor gene).39,40 However, somatic mutations of p53 are not commonly found in sporadic ependymomas and likely do not play a large role in their pathogenesis.41,42 Scattered reports of patients with ependymoma and Turcot’s syndrome (brain tumors plus colonic neoplasia) are found in the literature, with some patients developing more than one ependymoma.43–45 These patients have germline mutations in the APC gene on chromosome 5 and an overactive Wnt signaling pathway. The role of the APC gene and Wnt signaling in sporadic ependymomas has not been elucidated. Spinal ependymomas have also been reported in the context of multiple endocrine neoplasia type I due to mutation of a tumor suppressor gene on chromosome 11q13.46,47 Other families with an increased incidence of ependymoma but no currently recognized familial tumor syndrome have also been reported.48–50 Epidemiology studies have shown an increased incidence of breast cancer in the family members of children with ependymoma.
Cancer Genetics
There is a large body of literature on the karyotypic changes found in adult and pediatric ependymomas. As a group, ependymomas studied by G-banding karyotype show a frequent loss of genetic material on chromosomes 22q, 6q, 9q, 17p, and 11q and show a gain of genetic material on chromosome 1q.38,40,51–56 Loss of heterozygosity on chromosome 22q is more common in adult ependymomas and intramedullary spinal ependymomas and less common in pediatric ependymomas.40 There is some evidence that a gain of genetic material on chromosome 1q may be an early event in the initiation of ependymoma.51
Ependymomas from different regions of the central nervous system (CNS) have distinct genetic profiles. There appear to be three genetic groups of childhood posterior fossa ependymomas. Ependymomas from very young children frequently show a balanced karyotype, with few or no regions of chromosomal gain or loss (no observed gains or losses).57,58 A second subset of posterior fossa ependymomas has few regions of gain or loss other than gain of chromosome 1q. A third group of posterior fossa tumors shows recurrent regions of copy number gain across several autosomes. It is unclear whether there is any clinical or prognostic significance to these three groups. Supratentorial ependymomas have different regions of gain and loss, and many show homozygous deletion of the INK4A locus on chromosome 9, which is also frequently deleted in glioblastoma multiforme.58 This key tumor suppressor locus plays a role in both TP53 and RB signaling. Recurrent tumors show more extensive karyotypic abnormalities than the original tumors; whether this is due to biologic progression or secondary to random DNA damage from radiation or chemotherapy is not clear.59 Myxopapillary ependymomas show a high number of cytogenetic abnormalities (averaging nine per tumor) compared with other ependymomas.60 The most common changes seen in cases of classic ependymoma are gains of chromosomal material on chromosome arms 1q or 9 and losses of 6q, 22, and the X chromosome.61 Overall, our knowledge of the genetics of ependymoma is limited by the resolution of the techniques used and the small number of tumors examined. Identification of specific genes and pathways for the development of targeted therapies will require larger studies done at higher resolution to differentiate driver genes from passenger genes.
Stem Cell Biology
It has been suggested that in many tumors only a small subset of tumor cells is capable of “regrowing” or recapitulating the tumor. These so-called cancer stem cells (CSCs) have been found in a number of tumor types, including ependymoma.58 Ependymoma CSCs grow as neurospheres, can differentiate along several different lineages, and can recapitulate ependymoma when transplanted into immune-deficient mice. Conversely, the non-CSCs from an ependymoma do not grow when xenografted. CSCs from ependymoma samples, but not medulloblastoma CSCs, express markers of radial glial cells (i.e., Blbp, RC2, Glast).58 These data suggest that ependymomas arise from radial glial cells, a type of CNS progenitor cell that is in fact the cell of origin of ependyma.62 Indeed, ependymomas from different regions of the nervous system have expression profiles similar to those of radial glial cells from those same regions, supporting a radial glial cell origin. A radial cell origin for ependymoma would certainly help explain the extraventricular location of many supratentorial ependymomas. Knowledge of the cell of origin may allow the creation of genetically modified mouse models of ependymoma by driving oncogene expression or the deletion of tumor suppressor genes in the radial glial cell compartment. These models would be welcome, because the shortage of model systems for ependymoma is undoubtedly responsible for the lack of research on the subject. There are no well-characterized ependymoma cell lines and no genetically modified mice that develop ependymoma; ependymoma xenografts have only recently been described.63
Ependymoma Variants
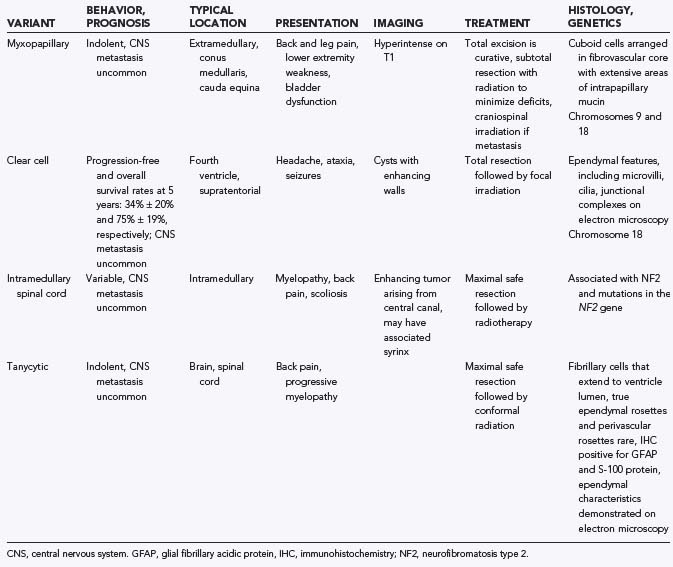
There are a number of histologic variants of ependymoma that have a distinct appearance under the light microscope and are likely separate entities from classic ependymoma, each with its own unique biology (Table 200-1).
Neuroimaging
Although most ependymomas enhance, they often contain areas that do not enhance; thus, careful scrutiny of the preoperative films is warranted to avoid missing tumor at the time of resection. It is important to compare the pre- and postoperative films side by side to determine the location and extent of residual tumor. Most children with recurrence or progression of tumor present between 12 and 24 months after the initiation of radiation therapy. Although there is no definitive proof, it is well accepted that early detection of recurrence maximizes the opportunity for salvage therapies. Some retrospective studies have even suggested that the early detection of asymptomatic recurrence of ependymoma on surveillance imaging leads to better outcomes than diagnosis when the recurrence becomes symptomatic.64,65
Initial Surgery
Children with ependymoma most often present with localized, invasive disease. Most recurrences are local, but subarachnoid dissemination occurs rarely and is usually fatal. The surgeon plays the most important role in the treatment of ependymoma, because surgery has the greatest impact on the quality and quantity of the patient’s future life. The overriding value of gross total resection of ependymoma has been demonstrated in several institutional retrospective reviews and two prospective phase III trials.2–5,7–9,16,19,27 Although complete resection is of paramount importance in the treatment of ependymoma, it is achieved in only 42% to 62% of patients.3,5,8,19 Complete resection is more readily achieved in supratentorial tumors and in those that arise from the roof of the fourth ventricle. Gross total resection of tumors that invade the floor of the fourth ventricle or pass out the foramen of Luschka to involve the lower cranial nerves is much more difficult, and surgical complication rates are higher.
Sutton and colleagues5 retrospectively analyzed 45 patients with ependymoma and found that the 5-year survival after gross total resection or near total resection was 60%, but with subtotal resection (defined in this chapter as <90% resection) it fell to 21%. Pollack and associates8 reported a 5-year survival of 80% after gross total resection, compared with 22% after less than total resection. Perilongo and coworkers4 retrospectively evaluated 92 children with ependymoma: the 10-year survival after gross total resection was 70%, and the progression-free survival estimate was 57%. With subtotal resection the 10-year survival was 32% and the 10-year progression-free survival was 11%. Robertson and colleagues9 prospectively treated 32 patients on the Children’s Cancer Group Protocol 921 and found that the 5-year progression-free survival was 66% for patients with less than 1.5 cm2 of residual tumor and 11% for those with more tumor. Although no randomized trial of complete versus partial resection for ependymoma has been (or will be) carried out, there is overwhelming evidence that cytoreductive surgery is beneficial to children with ependymoma. Children whose tumor burden can be decreased to minimal residual disease have the best chance at long-term survival.
Surgery without adjuvant treatment (radiation or chemotherapy) for children with ependymoma has been studied by two groups.17,66 The results of these studies demonstrate that surgery alone is probably a reasonable therapeutic option for children with supratentorial ependymoma in the absence of pathologic features of anaplastic ependymoma and when the surgeon is able to take a rim of white matter around the tumor. Biopsies of the walls of the resection cavity should be performed if surgery is contemplated as the sole treatment modality.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


