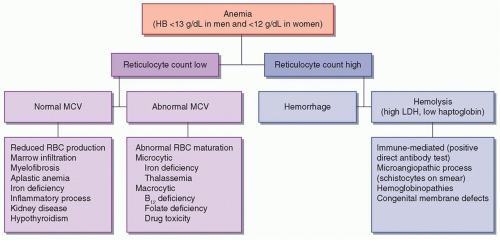
Anemia is defined as a hemoglobin (HB) concentration of less than 12 g/dL in women and 13 g/dL in men. There are numerous possible etiologies for anemia, which are presented in
Figure 118.1. Measurement of the reticulocyte count helps determine whether the bone marrow is responding “appropriately” to anemia—if so, it implies that the cause is hemorrhage or hemolysis; if not, there is a problem with RBC production. RBC indices, especially the mean cell volume (MCV), provide further information concerning the differential diagnosis. Microcytosis (MCV <80) implies that there is a cytoplasmic problem with the production of HB and the maturation of RBCs. Specific causes of microcytic anemia include iron deficiency and various hemoglobinopathies. Macrocytosis (MCV > 100) implies that there is a nuclear defect in developing RBCs, which may be due to vitamin B
12 and folate deficiency or drug toxicity. Normocytic anemia suggests that there is a pathologic bone marrow process or that there is reduced stimulation for RBC production. This section will focus on the neurologic implications of anemia and on sickle cell disease, which is a major cause of stroke in children and young adults.
Anemia and Transfusion in Hospitalized and Critically III Neurologic Patients
Anemia is one of the most common medical complications to be encountered among hospitalized patients, especially in the intensive care unit. The development of an HB concentration less than 10 g/dL has been reported in about 40% to 50% of neurocritical care patients with traumatic brain injury (TBI) or subarachnoid hemorrhage (SAH). The etiology of anemia in hospitalized patients is multifactorial. RBC production is impaired during systemic inflammation, as cytokines blunt the production of EPO and prevent progenitor cells from incorporating iron. RBC loss is accelerated by the need for frequent phlebotomy, reduced RBC survival, and (in some cases) hemorrhage. Hemodilution produced by administration of large volumes of intravenous fluids or phlebotomy may also contribute.
Optimal care of patients with acute brain injury and various forms of stroke involves the protection of salvageable brain tissue and the prevention of secondary injury. Reduced O2 delivery to ischemic penumbra may produce increasing neurologic damage. The amount of O2 reaching tissues is the product of local blood flow and arterial O2 content, which in turn is dependent on the HB concentration and the degree to which it is saturated with O2.
Anemia is initially well tolerated by most patients for several reasons: systemic O
2 delivery exceeds O
2 consumption by a large amount, tissues have the capacity to increase O
2 extraction in the
setting of reduced delivery, and sympathetic stimulation results in increased cardiac output. In the brain, the normal response to anemia is cerebral vasodilation, with a resulting increment in cerebral blood flow (CBF). Experiments in healthy volunteers demonstrate that neurocognitive impairment begins at an HB concentration below approximately 7 g/dL. It is likely that the HB threshold for neurologic deterioration is higher in brain-injured patients, in whom autoregulatory mechanisms may be impaired.
The development of anemia is associated with worsened outcomes in patients with TBI, SAH, and intracerebral hemorrhage (ICH). Studies using invasive multimodal neurologic monitoring have found that anemia is associated with lower brain tissue O2 tension and high cerebral lactate concentrations. Patients with SAH, in particular, are vulnerable to delayed cerebral ischemia. Administration of RBC transfusions improves O2 delivery to the brain and increases physiologic “reserve” in regions of the brain with a high O2 extraction fraction.
Historically, clinicians would give RBC transfusions to maintain HB concentrations greater than 9 to 10 g/dL in brain-injured patients. However, allogeneic RBC transfusions have potential adverse effects, including transfusion-related acute lung injury (TRALI) and immunosuppression with an increased risk of nosocomial infections. Randomized trials in general critical care patients have found no advantage, and possible harm, when RBC transfusions are used to maintain HB concentrations above 10 g/dL compared with a transfusion threshold of 7 g/dL [
Level 1].
1,
2 However, there were few brain-injured patients in these studies and HB concentrations of 7 to 9 g/dL may be too low in some brain-injured patients. There is substantial variability of practice, which will continue until clinical trials are performed specifically in brain-injured patients. Consensus guidelines in the setting of SAH recommend maintaining HB levels greater than 8 to 10 g/dL.
Sickle Cell Disease
Sickle cell disease (SCD) is a group of genetic disorders characterized by the presence of “sickle hemoglobin” (HBS) caused by a mutation in the β-globin gene, whereby the sixth amino acid changes from glutamic acid to valine. The most common and severe form is sickle cell anemia, which occurs in patients homozygous for the HBS allele. There are other SCD variants where HBS is inherited form one parent and another abnormal HB from the other. Stroke is the most common neurologic complication of SCD.
EPIDEMIOLOGY
The global burden of SCD is increasing, especially in Africa and India. About 8% of African-Americans are heterozygous for HBS and 1 in 600 are homozygous. Another 2% to 3% carry the HBC allele, which is attributable to a glutamic acid to lysine substitution. The incidence of stroke in children with SCD is about 300 to 400 times higher than the rate of stroke in other children, such that the chance of having a stroke by 20, 30, and 45 years of age is estimated to be about 11%, 15%, and 24%, respectively. The highest incidence occurs between the ages of 2 and 9 years. Intracranial hemorrhage is less common but increases in incidence between 20 and 30 years of age. Stroke is a major cause of premature death and disability in patients with SCD. Untreated, stroke may recur in as many as two-thirds of patients within 2 years. By far the strongest risk factor for stroke is a previous stroke or transient ischemic attack (TIA). Other risk factors include the degree to which HB is reduced, hypertension, frequent episodes of acute chest pain syndrome, leukocytosis, and lower pulse oximetry values.
Even in patients who have not developed overt stroke, magnetic resonance imaging (MRI) studies demonstrate that silent cerebral infarcts are common, occurring in more than a third of patients. Previous infarcts are seen especially in watershed regions and are associated with cognitive impairment and a higher subsequent risk of overt stroke. Areas of restricted diffusion, suggestive of recent cerebral ischemia, can be detected even when patients are asymptomatic, suggesting that patients are at constant risk. Identified risk factors for silent cerebral infarcts include lower baseline HB concentration, higher systolic blood pressure, and male gender.
PATHOBIOLOGY
O2 is normally transported by adult hemoglobin (HBA) consisting of two α and β polypeptide chains that encircle a heme moiety. Unlike HBA, HBS has a tendency to polymerize when it is deoxygenated, which in turn disrupts the normal architecture and flexibility of RBCs, causing them to take on sickle-shaped morphology. Sickling of RBCs interferes with their transit through capillaries and venules, causes them to adhere to endothelium, and increases blood viscosity, all of which may produce microvascular occlusion and tissue ischemia. Increased hemolysis occurs in the spleen. Most of the clinical manifestations of sickle cell anemia are attributable either to vaso-occlusion or hemolysis.
Multiple factors other than just stasis and sluggish microvascular flow are implicated in causing cerebral ischemia. Adherence of sickled RBCs to vascular endothelium induces a cascade of events that produce leukocyte recruitment, inflammation, intimal hyperplasia, fibrosis, and thrombosis. Intravascular hemolysis and release of free HB scavenges nitric oxide, the production of which may also be impaired by endothelial damage, thereby interfering with maintenance of normal vascular tone. These factors contribute to impaired CBF autoregulation, making the brain vulnerable both to hyperemia and ischemia.
Imaging studies with digital subtraction or magnetic resonance (MR) angiography demonstrate that many patients have various forms of vasculopathy. By far the most common cerebrovascular abnormality is stenosis of proximal intracranial vessels, especially in the anterior circulation, which may be accompanied by relative hypoperfusion if perfusion imaging is performed. Involvement of extracranial vessels is less common but does occur and may lead to cervical artery dissection and/or embolic strokes. Over time, with persistent severe intracranial stenosis, there may be development of collateral vessels resembling those of Moyamoya disease. The presence of such collaterals is a marker of a greater degree of vasculopathy and has been identified as a major risk factor for future stroke. These friable vessels are also vulnerable to bleeding. Intracranial aneurysms may develop in unusual locations with a possible predilection for the posterior circulation.
CLINICAL MANIFESTATIONS
SCD patients with cerebrovascular disease present most often with TIA or ischemic stroke. Among those with hemorrhage, SAH is more common than ICH or intraventricular hemorrhage. Repeated silent infarcts cause progressive neurologic deterioration. Children in whom previous infarcts are detected on a MRI scan have, on average, lower performance on neurocognitive testing and worse school performance. They are also more likely to have psychological concerns, such as anxiety and depression. Similar observations have been made in adults with sickle cell anemia, in whom the degree of anemia is associated with more severely impaired cognitive function. Neuroimaging reveals that patients with SCD have
a greater degree of thinning of the frontal lobe cortex, as well as reduced basal ganglia and thalamus volumes.
SCD may rarely cause complications in the spinal cord and peripheral nervous system. There have been numerous case reports of spinal cord infarction, most often involving the cervical cord and causing quadriparesis. Ischemic injury of peripheral nerves presents as mononeuritis multiplex. Functional asplenia is a well-recognized complication of SCD. The resulting immunosuppression predisposes to bacterial infections, including meningitis, with the risk being high enough to justify prophylactic penicillin usage until at least the age of 5 years. Fever should therefore be considered a medical emergency.