Fig. 19.1
Multihormonal microadenoma in the MEN1 pancreas immunostained for (a) glucagon and (b) pancreatic polypeptide
In MEN1 patients, all somatic cells harbor a heterozygous germline mutation of the MEN1 tumor suppressor gene. By combining fluorescence in situ hybridization of the MEN1 locus at 11q13 and the centromeric region of chromosome 11q with hormone immunostaining, it was demonstrated that microadenomas, defined as compact neuroendocrine cell aggregates lacking the typical islet architecture and expressing usually only one hormone, show loss of heterozygosity (LOH) at 11q13 and thus lack one MEN1 allele which characterizes the neoplastic stage in MEN1 [11]. Normal islet cells were found to retain both MEN1 alleles. Interestingly, a few islets were identified that exhibited glucagon cell hyperplasia compared to the number of insulin cells in the same islets. As these glucagon cells also hold a central position in the islets, these intraislet glucagon cell clusters were regarded as precursors to microadenomas. The glucagon cells in these lesions either still retained both MEN1 alleles or already showed LOH at 11q [11] (Fig. 19.2). This suggested that the hyperplastic glucagon cells, that carry, like all cells of the body, the MEN1 germline mutation on one allele, were in a transient stage. While some had not yet assumed the neoplastic genotype, others had already performed the transition from the non-neoplastic to the neoplastic stage characterized by the allelic loss of 11q13. We do not know what mechanisms and factors force some islet cells, in particular glucagon cells, into proliferation and produce hyperplastic changes, but this process could be related to an increased responsiveness of the glucagon cell in the setting of MEN1 to certain growth factors or some other growth-enhancing mechanisms. The glucagon cells obviously share this ability with gastrin cells in the duodenal mucosa where focal gastrin cell hyperplasias were identified as precursors to gastrin microadenomas [12].
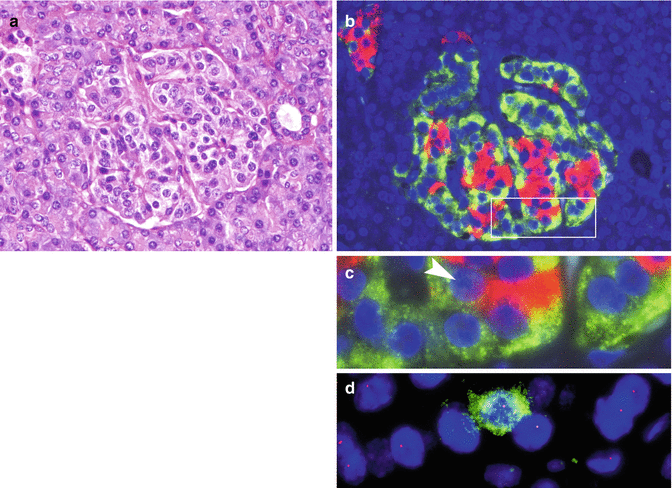
Fig. 19.2
Islet with glucagon cell hyperplasia turning into monoclonal cell proliferation. (a) H&E-stained islet with almost normal structure. (b) Serial section showing glucagon cell hyperplasia (immunofluorescence: glucagon in green, insulin in red). (c) Enlarged box-labeled area in b. Arrow head indicates the insulin cell marked in green in panel d. (d) Retention of heterozygosity of 11q13 in a single insulin cell (green) and LOH on 11q13 in unstained glucagon cells (From Perren et al. [11] with permission of publisher)
These results implicate first that the allelic loss at 11q13 in an islet cell sets the stage for NET development; second, it seems that the glucagon cell is the islet cell type that is most responsive to hyperplastic and finally neoplastic stimuli. The reason why glucagon cells are particularly prone to these changes is yet not known.
Of the many microadenomas that are developing in the MEN1 pancreas, apparently only a small fraction proceeds to macrotumors (>0.5 cm and larger). This selective tumor evolution seems to be related to additional genetic alteration. When MEN1 microadenomas and macrotumors were analyzed for ATRX/DAXX mutations that correlate with alternate lengthening of telomeres and chromosomal instability [13], and are commonly found in advanced sporadic PanNETs [13, 14], microadenomas lacked the ATRX/DAXX mutations, while advanced macrotumors carried them [15]. This suggests that ATRX and DAXX mutations are late events in the tumorigenesis of pancreatic neuroendocrine neoplasms, and only those microadenomas may progress to macrotumors that acquire these genetic alterations.
19.3 Glucagon Cell Adenomatosis (GCA)
Recently, GCA has been described as a distinct multicentric neoplastic disease of the pancreas that is preceded by glucagon cell hyperplasia of the islets [16, 17] (Fig. 19.3). There may be earlier reports on GCA (see [7]), but the descriptions of the changes are often difficult to classify. GCA is a disease that involves the entire pancreas, except for the so-called PP lobe in the head of the pancreas, where glucagon cells are infrequent [17a]. In the remaining pancreas, the islets are looking normal, when they are small. The bigger islets, however, display glucagon cell hyperplasia that correlates in its extent with the islet diameter (Fig. 19.3). The largest islets then imperceptibly transform into glucagon cell microadenomas (Fig. 19.4). Rarely, the tumors may also contain a considerable number of PP cells. In the most advanced cases, there are single macrotumors (5–20 mm in size) (Fig. 19.5), some of them with cystic changes, as it is typical for glucagon-producing tumors [18]. All tumors were well differentiated, with a Ki67 index below 2 %. However, despite these benign features, lymph node micrometastases were detected in one patient. Clinically, the patients who were tested were found to have elevated glucagon serum levels, but usually presented without signs of a glucagonoma syndrome [7, 16].
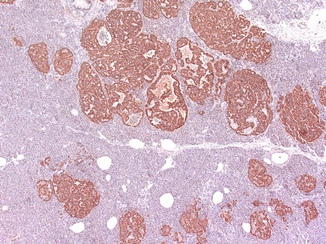
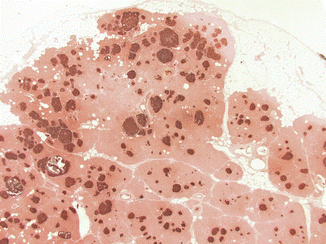
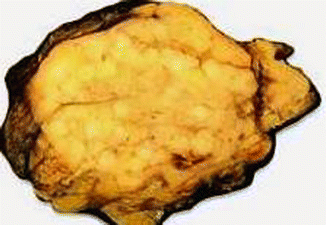
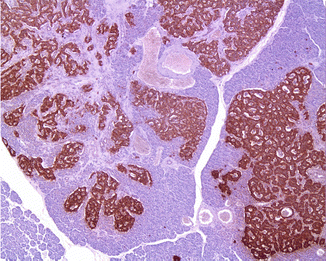
Fig. 19.3
Glucagon cell adenomatosis: islets with glucagon cell hyperplasia (lower part) next to multiple small glucagon-positive microadenomas (upper part of the illustration)
Fig. 19.4
Glucagon cell adenomatosis: immunostaining for glucagon shows islets with glucagon cell hyperplasia that imperceptibly transforms into microadenomas
Fig. 19.5
Glucagon cell adenomatosis: cut surface of a pancreatic resection specimen shows multiple whitish small tumors
Genetically, the disease was found to be unrelated to MEN1 and VHL. In half of the six patients of our GCA series, however, it was associated with inactivating homozygous and double-heterozygous germline mutations of the glucagon receptor (GCGR) gene, that were probably inherited in an autosomal, recessive mode as demonstrated in the parents of one of our patients [17a]. This suggests that a single heterozygous mutation of the GCGR gene probably does not cause the disease.
Analysis of the GCGR gene alterations suggests that the mutations cause a loss of receptor function, an assumption which is also supported by murine models [19], in which the deletion of the GCGR gene results in glucagon cell hyperplasia and finally also neoplasia [20]. While a dysfunctioning GCGR nicely explains the absence of a glucagonoma syndrome in the presence of elevated glucagon levels in the serum, the mechanisms that lead to the proliferative glucagon cell changes are unclear. It can be, however, postulated that there is a factor that stimulates the growth of glucagon cells. Since gluconeogenesis in the liver particularly depends on the effect of glucagon and thus on a functioning GCGR, it might be that the liver is the source of the postulated factor which is secreted in order to increase glucagon production by increasing glucagon cell number.
The mechanisms that lead to GCA in the patients of our series, in whom no GCGR mutations were detected, are also not known. However, the fact that GCA seems to occur without GCGR mutation, as has also been described in a recent case report on GCA [21], implies that probably alternate mechanisms exist that indirectly affect the GCGR signaling. GCA seems therefore to be a heterogeneous disease. If the patients with and without GCGR mutations are compared with each other, it appears that the mutation-positive patients have a more severe disease in terms of tumor size and number than the mutation-negative patients [17a].
If disturbed GCGR signaling, either directly or via an alternate pathway, plays an important role in the initiation of the disease, the question that has to be addressed next is what stimulates the development of the subsequent neoplastic changes. Most likely, additional genetic changes are required for this development, but so far, no findings are available explaining this second step in the tumorigenesis of GCA. Since in rare cases GCA may cause lymph node metastases, its name has perhaps to be changed in the future to glucagon cell hyperplasia and neoplasia.
19.4 Insulinomatosis
Insulinomatosis has been described as a distinct neoplastic disease of the pancreas, in which, syn- and metachronously, multiple small and single large insulin-producing tumors develop [22] (Fig. 19.6). The affected patients present with hyperinsulinemic hypoglycemia which, after removal of the large visible insulinoma(s), has a great risk of recurrence because of the development of new large tumors in the remaining pancreas. The patients therefore often have a history of repeated pancreatic surgery that finally results in total removal of the pancreas. It seems that the tumors are not preceded by an insulin cell hyperplasia in the islets, but rather originate from compact insulin cell clusters, often with a trabecular pattern, devoid of any other islet cell type. In all but one patient of our series, the disease followed a benign course. The patient with malignant disease developed lymph node and liver metastases. So far, no genetic defect has been discovered, but familiarity may occur.
 Endocrinological Approach to the Diagnosis of Pancreatic Neuroendocrine Neoplasms
Molecular Pathology of Pancreatic Neuroendocrine Neoplasms
The Role of Nuclear Medicine in the Diagnosis of Pancreatic Neuroendocrine Neoplasms
Immunohistochemical Approach to the Diagnosis and Prognostic Evaluation of Pancreatic Neuroendocrine Neoplasms
Cytological Diagnosis of Pancreatic Neuroendocrine Neoplasms
Mixed Adenoneuroendocrine Carcinoma of the Pancreas
Endocrinological Approach to the Diagnosis of Pancreatic Neuroendocrine Neoplasms
Molecular Pathology of Pancreatic Neuroendocrine Neoplasms
The Role of Nuclear Medicine in the Diagnosis of Pancreatic Neuroendocrine Neoplasms
Immunohistochemical Approach to the Diagnosis and Prognostic Evaluation of Pancreatic Neuroendocrine Neoplasms
Cytological Diagnosis of Pancreatic Neuroendocrine Neoplasms
Mixed Adenoneuroendocrine Carcinoma of the Pancreas




Related posts:
Endocrinological Approach to the Diagnosis of Pancreatic Neuroendocrine Neoplasms
Molecular Pathology of Pancreatic Neuroendocrine Neoplasms
The Role of Nuclear Medicine in the Diagnosis of Pancreatic Neuroendocrine Neoplasms
Immunohistochemical Approach to the Diagnosis and Prognostic Evaluation of Pancreatic Neuroendocrine Neoplasms
Cytological Diagnosis of Pancreatic Neuroendocrine Neoplasms
Mixed Adenoneuroendocrine Carcinoma of the Pancreas
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
