Hypoxic-Ischemic Encephalopathy
Alexandra S. Reynolds
Sachin Agarwal
INTRODUCTION
Hypoxic-ischemic encephalopathy (HIE) is a term used to describe cerebral dysfunction after a global insult to the brain. Mechanisms of such damage include prolonged interruption of blood flow and/or oxygen, most commonly after cardiac arrest.
EPIDEMIOLOGY
Approximately 630,000 cardiac arrests occur in the United States per year with an overall 14% survival to discharge. Despite an increased survival from all-rhythm cardiac arrest, the mortality is at least 50% in those who have survived to the hospital admission and overall disease-specific mortality is around 90%. Brain injury alone is the primary cause in 68% of patients. Among all out-ofhospital cardiac arrest patients between 2000 and 2006, patients who found in asystolic arrest had the worst outcomes (1% survival to discharge and 0.5% 30-day survival), whereas those with shockable initial rhythm (ventricular fibrillation [VF] or pulseless ventricular tachycardia) had the best outcomes (15%-20% survival to discharge). Those with pulseless electrical activity (PEA) fell somewhere in the middle (5%-8% survival to discharge). In general, the faster the return to spontaneous circulation (ROSC), the better the outcome.
These numbers do not capture the neurologic morbidity associated with cardiac arrest. Those who survive may still have significant neurologic deficits including personality changes and problems with memory. Further, a proportion of patients will ultimately remain in minimally conscious or persistent vegetative states. The true proportions are unknown because of high rates of withdrawal of care in patients who remain comatose after a cardiac arrest leading to a “self-fulfilling prophecy.”
PATHOBIOLOGY: CAUSES OF HYPOXIC-ISCHEMIC ENCEPHALOPATHY
CARDIAC ARREST
Cardiopulmonary arrest is a complex process that causes diffuse damage to the brain. The initial mechanisms of damage include hypoxemia and hypoperfusion due to circulatory arrest. However, as the cardiac arrest progresses, damage is caused by resultant hypoglycemia, acidosis, and toxin accumulation (see following text). The model of cardiac arrest resuscitation involves three time-sensitive ischemic-reperfusion phases, including electrical, circulatory, and metabolic. Although rapid reperfusion is needed after ischemia, it can also paradoxically contribute to tissue injury and destruction. Various mechanisms of cell death, including necrosis, apoptosis, and autophagy-associated cell death, have been implicated. Tissue necrosis results in swelling and this is exacerbated by rebound hyperemia from reperfusion as well as progression of the inflammatory cascade. On a molecular level, reintroduction of glucose causes formation of nitric oxide and oxygen free radicals that can induce DNA damage and consumption of NAD + leading to a second round of cell death. Studies involving cellular, animal, and human models suggest that therapeutic hypothermia (TH) has the potential to mitigate these deleterious processes (Table 37.1) even after a period of ischemia. Models where hypothermia was initiated before reperfusion have resulted in the most cellular protection.
PROLONGED HYPOTENSION
Hypotension results in preferential injury to parts of the central nervous system that lie in between vascular distributions. Areas supplied by narrowed arteries are also at risk for hypoperfusion in low-flow states like carotid stenosis or in heart failure with low cardiac output. Low cerebral blood flow results in patchy ischemic infarctions. Watershed areas in the brain affected by hypotension include the cortical border zones between the anterior cerebral and middle cerebral arteries (ACA-MCA) and middle cerebral and posterior cerebral arteries (MCA-PCA) or internal border zone areas in the periventricular white matter or the deep structures supplied by the recurrent artery of Heubner and the anterior choroidal and
lenticulostriate arteries. In the spinal cord, the anterior portion of the midthoracic distribution is considered to be the watershed region; however, one study showed disproportionate damage to the lumbosacral spinal cord after prolonged hypotension and/or cardiac arrest.
lenticulostriate arteries. In the spinal cord, the anterior portion of the midthoracic distribution is considered to be the watershed region; however, one study showed disproportionate damage to the lumbosacral spinal cord after prolonged hypotension and/or cardiac arrest.
TABLE 37.1 Mechanisms of Neuroprotection with Therapeutic Hypothermia | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
HYPOXIA
Pure hypoxic hypoxia can occur in the absence of circulatory breakdown, in the case of aspiration, hanging/strangulation, or drowning. Hypoxic damage to the brain occurs solely due to decreased partial pressure of blood oxygen and results in a lesser degree of injury than combined hypoxic-ischemia. However, prolonged hypoxia will progress to cardiac arrest. Once oxygen supply to the brain is interrupted, adenosine triphosphate (ATP) stores only last for minutes. After energy-dependent membrane ion transport starts to fail, damaged cells release excitotoxic glutamate, intracellular calcium begins to accumulate and activates proteases and phospholipases, and damage is propagated via inflammatory cascades. Intracellular influx of calcium in surrounding areas leads to diffuse cell death. The areas particularly susceptible to hypoxic damage are hippocampal CA1 neurons, thalami, cerebellar Purkinje cells, and cortical pyramidal cells in layers 3, 5, and 6. Among the cortical areas, perirolandic areas are most vulnerable because of the high concentration of N-methyl-D-aspartate (NMDA) receptors. White matter injury also occurs because of damage to oligodendroglial cells.
CARBON MONOXIDE POISONING
Carbon monoxide (CO) primarily exerts harmful effects to the brain by causing hypoxemia in a so-called anemic-histotoxic hypoxia. As in hypoxic hypoxia, preservation of circulation portends a lesser damage to the brain. CO preferentially binds to heme proteins and displaces bound oxygen because of its greater affinity for hemoglobin. CO thus limits oxygen-carrying capacity of blood and impairs mitochondrial function of the tissues attempting to use oxygen, resulting in a hypoxic and acidotic state. Ultimately, CO poisoning causes intravascular and intracellular oxidative stress, inflammation, and apoptosis. The cortical laminae and basal ganglia are particularly prone to damage. Additional reports of delayed demyelination in the centrum semiovale have been published. In severe cases, cardiac toxicity can occur resulting in hypotensive brain injury as well.
CYANIDE POISONING
Cyanide poisoning causes metabolic failure by inhibiting oxidative phosphorylation. It results in direct metabolic damage to the brain but ultimately causes mixed injury due to early cardiac failure. Cyanide impairs oxidative phosphorylation by inhibiting the mitochondrial cytochrome oxidase a3 complex. Mitochondria are unable to produce ATP and increased glycolysis occurs, quickly resulting in a metabolic acidosis as pyruvate is reduced to lactate. Coronary artery vasoconstriction occurs and cardiac output decreases suddenly, causing hypotension and cardiac shock. The brain is damaged not only from this hypotension but also from metabolic failure on a cellular level.
PROFOUND HYPOGLYCEMIA
Glucose is the primary source of energy for the brain in both adults and in infants, and prolonged or repeated hypoglycemic episodes can therefore cause significant injury to the brain. Concurrent hypoglycemia also exacerbates the damage caused by hypoxemia. Brain damage usually occurs once serum glucose levels fall below 20 mg/dL. Mechanisms of cell death with hypoglycemia are similar to hypoxia, including excitotoxic glutamate and aspartate release and involvement of NMDA receptors. More recent studies have shown that activation of poly (ADP ribose) polymerase-1 (PARP-1), release of zinc from nerve terminals, and activation of calciumdependent protease calpain also contribute to neuronal death. Further, depolarization of the mitochondrial membrane leads to apoptosis. The parts of the brain most sensitive to low-glucose levels are the hippocampus (CA1, CA3, and dentate gyrus), caudate, putamen, and insular cortex.
Damage from hypoxic-ischemic injury also changes as the brain matures. In neonates, HIE often results in parietooccipital and thalamic damage, whereas in infants, basal ganglia damage is more frequent and in children, parietotemporal changes are seen.
CLINICAL FEATURES
The clinical manifestations of HIE can range from mild changes in cognition to coma. Damage to the hippocampus can result in memory deficits. Damage to the basal ganglia can present as a variety of movement disorders. Thalamic damage can result in a wide variety of symptoms from sensory changes to dementia or coma. Damage to the Purkinje cells results in cerebellar dysfunction. Cortical damage can result in inattentiveness, executive dysfunction, or aphasia. Because of the resistance of the deep gray matter of the brain stem to anoxic injury compared with the neocortex, HIE often results in coma with preservation of brain stem reflexes. However, brain death can occur with prolonged hypoxic-ischemic injury.
Watershed injury in the brain can manifest as proximal greater than distal extremity weakness and ultimately spasticity (“man-ina-barrel syndrome”). Watershed injury to the spinal cord results in an “anterior cord syndrome” or Beck syndrome, including motor paralysis below the level of the lesion, loss of pain and temperature but intact proprioception and vibratory sensation, and loss of bowel and/or bladder control.
Lance-Adams syndrome, or posthypoxic action myoclonus, can occur several days after recovery from hypoxic injury to the brain.
The rare syndrome of delayed posthypoxic leukoencephalopathy (DPHL) following seemingly full recovery from a comatose state can occur days to weeks after the initial insult. The onset is usually insidious and can manifest as personality changes, irritability, inattentiveness, confusion, and memory problems. This can progress to include extrapyramidal motor symptoms and, in some cases, eventually coma or death. Those patients who survive stabilize to one of two ultimate syndromes: either parkinsonism with hallucinations or odd behaviors, or akinetic mutism. DPHL may occur as a result of any cause of HIE mentioned earlier but is most often seen in cases of acute CO poisoning or opioid overdose. The main histopathologic patterns seen in this syndrome are demyelination in the centrum semiovale and necrosis of the cortical laminae and basal ganglia, which correlate with brain magnetic resonance imaging (MRI) features. Severity and persistence of DPHL sequelae have been correlated to the presence and persistence of low signal intensity on the apparent diffusion coefficient (ADC) map on cranial MRI.
DIAGNOSIS
IMAGING
CT head is usually normal immediately after hypoxic-ischemic insult. In severe injury, there can be evidence of loss of the gray-white junction within hours (Fig. 37.1A). After 24 to 48 hours, linear
hyperdensities outlining the cortex develop, corresponding to laminar necrosis of the cortex. After days, the “reversal sign” develops where cerebral white matter appears hyperdense than cortical gray matter. Pseudosubarachnoid hemorrhage can also occur when the falx cerebri and tentorium cerebelli appear hyperdense as compared to the adjacent injured parenchyma (see Fig. 37.1B).
hyperdensities outlining the cortex develop, corresponding to laminar necrosis of the cortex. After days, the “reversal sign” develops where cerebral white matter appears hyperdense than cortical gray matter. Pseudosubarachnoid hemorrhage can also occur when the falx cerebri and tentorium cerebelli appear hyperdense as compared to the adjacent injured parenchyma (see Fig. 37.1B).
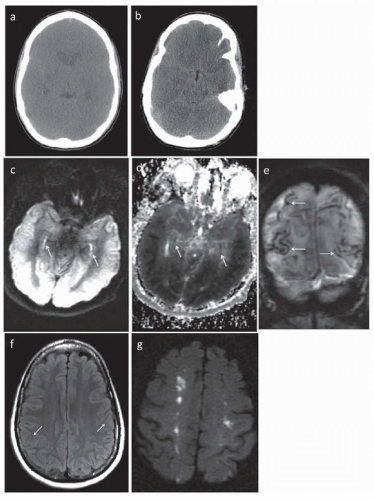 FIGURE 37.1 Imaging findings after HIE. CT scan done (A) 12 hours and (B) 10 days after a neardrowning episode. A: There is diffuse loss of gray-white matter differentiation and severe global cerebral edema. There are bilateral, but right greater than left, hypodensities of the basal ganglia. B: The diffusely attenuated white and gray matter result in a pseudosubarachnoid hemorrhage appearance of the relatively hyperdense dura. DWI (C) and ADC (D) sequences of an MRI done 2 days after a primary respiratory arrest showing restricted diffusion in the bilateral hippocampi (arrows) and diffusely throughout the cortex. E: Susceptibilityweighted imaging (SWI) sequence in coronal section done 7 days after a PEA arrest showing areas of laminar necrosis in the posterior parietal lobes (arrows). F: FLAIR sequence 10 days after cardiopulmonary arrest only showing mild swelling and indistinctness of the gray-white junction in the bilateral parietal lobes (arrows). G: DWI sequence of an MRI done 11 days after a PEA arrest resulting from a type A aortic dissection. There are scattered foci of restricted diffusion along the ACA-MCA and MCA-PCA border zones, more prominent on the right, consistent with watershed infarct. |
MRI brain is the earliest way to visualize damage, as early cytotoxic edema can be visualized on diffusion-weighted imaging (DWI) and can be quantified using the ADC (see Fig. 37.1C-G). Importantly, DWI and ADC changes may not be apparent early on and thus early imaging may underestimate the injury burden. Moreover, there may be attenuation of ADC values for every 1°C decrease in body temperature. DWI sequences can pseudonormalize by seven days but T2 hyperintensities on fluid-attenuated inversion recovery (FLAIR) sequences develop after the first week. The damage is most often symmetric and corresponds in location to the neuronal subtypes that are preferentially damaged (see pathobiology section). With time, focal laminar necrosis can be seen due to deposition of blood products (see Fig. 37.1E). In one small study, changes occurring over time within the same patient were analyzed. The location of the injury shifts with time from the initial hypoxic-ischemic event, starting in the cerebellum and basal ganglia in days 1 and 2, moving to the cortex around days 3 to 5, and involving subcortical white matter on days 6 to 12. Pronounced hippocampal abnormalities on DWI—the so-called bright hippocampus sign—has been associated with extremely poor outcomes (see Fig. 37.1C,D). In the case of DPHL, MRI findings are pathognomonic, with diffuse T2 hyperintensities predominantly in the dorsal frontal and parietal lobes.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


