Chapter 53D Infections of the Nervous System
Prion Diseases
Prion (pronounced pree-ahn) diseases are a group of uniformly fatal neurodegenerative diseases caused by the transformation of an endogenous protein, PrP (prion-related protein), into an abnormal conformation called the prion. The term prion is derived from the term proteinaceous infectious particle and was named by Stanley Prusiner, who discovered the protein (Prusiner, 1998). For many years, prion diseases were mistakenly thought to be due to “slow viruses,” in part owing to the transmissibility of the diseases and the long incubation period between exposure and symptom onset (Brown et al., 1986b; Gajdusek, 1977). Research by Stanley Prusiner and others, however, determined that the infectious agent did not contain nucleic acid, a component of viruses. Furthermore, treating prion-contaminated material with methods that inactivated viruses and other microorganisms did not prevent these diseases from being experimentally transmitted; yet methods that denatured or destroyed proteins prevented transmission, strongly supporting the theory that the causative agent was a protein (Gajdusek et al., 1977; Prusiner, 1982). The identification of the gene encoding human PrP (Oesch et al., 1985), PRNP, and mutations of this gene in patients with familial prion disease (Goldgaber et al., 1989; Hsiao et al., 1989) further helped support the prion hypothesis. In 1997, Stanley B. Prusiner received the Nobel Prize in Physiology and Medicine for his work on identifying the prion (Prusiner, 1998). Through animal models, identification of prion gene mutations causing prion disease in humans, and in vitro production of prions with transmissibility, it essentially has been proven that the prion protein is necessary and sufficient to cause prion disease (Kim et al., 2010; Makarava et al., 2010; Mead, 2006). Although prion diseases occur in animals and humans, this chapter will focus on human prion diseases and only discuss prion diseases in animals relevant to humans.
Human Prion Diseases
Perhaps one reason many find prion diseases so fascinating is that they are unique in medicine because they can occur in three ways in humans: spontaneously (sporadic), genetically, and through transmission (acquired) (Prusiner, 1998). Approximately 85% are sporadic, 15% are genetic, and fewer than 1% are acquired (e.g., iatrogenic). Sporadic prion disease, or sporadic Jakob-Creutzfeldt disease (sCJD), is thought to occur spontaneously. Genetic prion diseases (gPrD) are due to a mutation in the PRNP gene that encodes the prion protein, PrP; gPrDs are usually classified into three clinicopathological forms: familial CJD (fCJD), Gerstmann-Sträussler-Scheinker disease (GSS), and fatal familial insomnia (FFI). Although acquired prion diseases are the least common form of human prion disease, they are perhaps the most notorious, in part owing to their occurrence through inadvertent transmission of prions from animals to humans and from human to human. Because the genetic and acquired forms of human prion disease are less common, they will be discussed in less detail in this chapter than the much more common form, sCJD.
Epidemiology
The incidence of human prion diseases is about 1 to 1.5 per million per year in most developed countries, with some variability from year to year and between countries. This means annually there are about 6000 human prion cases worldwide and about 250 to 400 in the United States. The peak age of onset of sCJD occurs around a unimodal relatively narrow peak of about 68 years (Brown et al., 1986a). Because sCJD tends to occur within a relatively narrow age range, a person’s lifetime risk of dying from sCJD is estimated to be about 1 in 9000, much higher than the incidence (which is across all age groups) of 1 in a million. It is not clear why some countries such as Switzerland have a slightly higher incidence of about 1.9 per million per year, but this might be due to ascertainment bias, where in a small country it might be easier to track all cases.
History of Creutzfeldt-Jakob Disease Nomenclature
The history of the nomenclature for CJD is quite interesting. In 1921 and 1923, Alfons Jakob published four papers describing five unusual cases of rapidly progressive dementia. He stated that his cases were nearly identical to a case described earlier by his professor Hans Creutzfeldt in 1920. This disease was referred to for many decades as Jakob’s or Jakob-Creutzfeldt disease until Clarence J. Gibbs, a prominent researcher in the field, started using the term Creutzfeldt-Jakob disease because the acronym was closer to his own initials (Gibbs, 1992). It turns out that the cases Jakob described were very different than Creutzfeldt’s case, and that only two of Jakob’s five cases actually had the disease that we now call CJD (prion disease), whereas Creutzfeldt’s case did not (Katscher, 1998). Therefore, the name for prion disease should be Jakob’s disease or possibly Jakob-Creutzfeldt disease. But because the term JC disease (JCD) might be confused with progressive multifocal leukoencephalopathy (PML) caused by the JC virus, unfortunately we continue to use the term CJD. In this chapter, we use the terms Creutzfeldt-Jakob disease and CJD. Prion diseases also have been historically called transmissible spongiform encephalopathies (TSEs) because of two common properties of prion disease; because some gPrDs might not be transmissible, and not all human prion diseases have spongiform changes (now called vacuolation due to fluid-filled vesicles in the dendrites) on pathology, the term TSE will not be used in this chapter.
What are Prions?
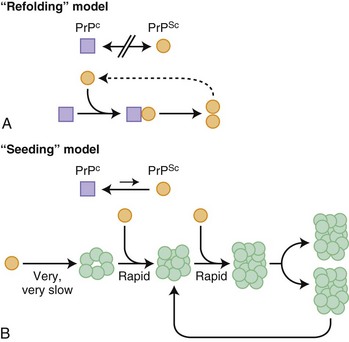
Before further discussion of various forms of prion diseases, it is important to understand what a prion is. Just as the nomenclature for human prion diseases is complicated, so is the terminology for the biology of prions. The normal prion protein (PrP) is referred to as PrPC, in which the C stands for the normal cellular form. Prion proteins, PrPC, can be transformed into prions, an abnormal infectious form of PrP called PrPSc, in which Sc refers to the abnormally shaped PrP found in scrapie, the prion disease of sheep and goats. PrPC and PrPSc essentially have identical amino acid sequences (except in genetic prion diseases; see later) but have different three-dimensional structures. Prions are characterized by their infectious properties and by the intrinsic ability of their structures to act as a template and convert the normal physiological PrPC into the pathological disease-causing form, PrPSc. Per the current prion model, when PrPC comes in contact with PrPSc, PrPC changes shape into that of PrPSc. PrPSc acts as a template for the misfolding of PrPC into PrPSc. It is believed that it is the accumulation of prions, PrPSc, in the brain that leads to nerve cell injury and death (Prusiner, 1998; Prusiner and Bosque, 2001), although some believe that it is not the accumulation of PrPSc, but rather the transformation of PrPC into PrPSc that causes neuronal injury and subsequent disease (Mallucci et al., 2007). How prions spread throughout the brain is not known. At least two models have been proposed, a refolding and a seeding model, which are not mutually exclusive (Fig. 53D.1).
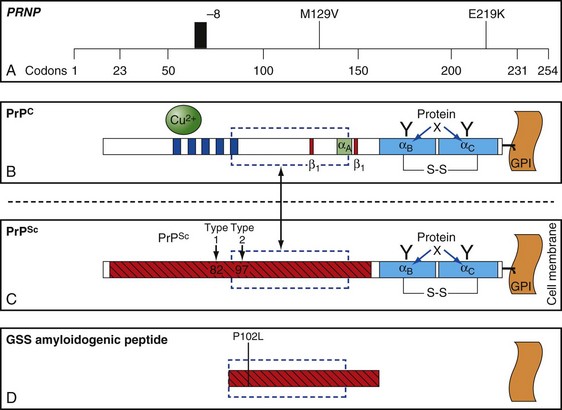
The function of PrP is still not completely known (Geschwind and Legname, 2008). It is evolutionary conserved, so it probably plays an important role in neuronal development and function (Kanaani et al., 2005). In humans, it is encoded by the PRNP gene located on the short arm of chromosome 20 (Basler et al., 1986; Oesch et al., 1985). PrPC is primarily membrane bound and resides primarily on nerve cell membranes and on other cells in the body including lymphocytes. The mature PrPC protein is attached to the outer cell membrane by a glycosylphosphatidylinositol (GPI) anchor (Borchelt et al., 1992, 1993; Taraboulos et al., 1992) (Fig. 53D.2). Mice that have had both copies of the open reading frame (ORF) of their PrP gene, Prnp, deleted (PrP−/−) have a normal lifespan and appearance (Bueler et al., 1992; Manson et al., 1994). Although clinically asymptomatic, they develop peripheral nerve demyelination (Nishida et al., 1999), have increased susceptibility to ischemic brain injury (Spudich et al., 2005; Weise et al., 2006), altered sleep and circadian rhythm (Tobler et al., 1997), altered hippocampal neuropathology and physiology including deficits in hippocampal-dependent spatial learning and hippocampal synaptic plasticity (Colling et al., 1997; Criado et al., 2005), and olfactory dysfunction (Le Pichon et al., 2008). When a slightly larger region beyond the ORF is deleted, mice develop ataxia and Purkinje cell loss later in life (Katamine et al., 1998; Rossi et al., 2001; Sakaguchi et al., 1996). Furthermore, conditional knockout mice in which the gene is not removed until after the mouse has already developed also appear normal and unaffected by gene removal.
PrPC binds to many proteins and cellular constituents. Animal and cell models have generated a variety of possible functions of PrPC including cell signaling, adhesion, proliferation, differentiation, and growth. Over the past year alone, there has been a dramatic increase in research studies postulating new roles for PrPC, including mice studies showing it mediates the toxic effects of the major protein in Alzheimer disease (AD), β-amyloid, and that it might be required for memory deficits in AD (Nygaard and Strittmatter, 2009). In fact, infusion of anti- PrPC antibodies have been used to treat cognitive deficits in AD mouse models (Chung et al., 2010; Gimbel et al., 2010; Gunther and Strittmatter, 2010). Importantly, mice devoid of PrPC can neither be infected with nor replicate prions, providing strong evidence that PrPC is necessary for prion disease (Bueler et al., 1993; Katamine et al., 1998; Prusiner et al., 1993).
Clinical Aspects of Human Prion Diseases
Sporadic Prion Disease
Sporadic CJD (sCJD) is also called classic CJD or sometimes simply CJD. It is thought to occur spontaneously, either through the spontaneous transformation of the prion protein, PrPC, into PrPSc or through a somatic mutation that results in the formation of a prion protein that is more susceptible to changing into PrPSc (see Watts et al., 2006, for a discussion on possible origins of sCJD). Nomenclature for prion diseases can be confusing. The term CJD is sometimes used to refer to all human prion diseases and sometimes to just the classic or sporadic form, sCJD. In this chapter, CJD will refer to all human prion diseases, whereas sCJD will be used to refer only to sporadic CJD.
Sporadic CJD is typically a very rapid disease with a median survival of about 7 to 8 months and mean survival of 4 months. More than 90% of patients are dead within 1 year of onset of symptoms. The mean age of onset is 68, and the median age is in the early 60s, although the range is from the 20s to 80s (Table 53D.1). Occurrence of sCJD at young (20s-40s) or old (>75) ages is uncommon (Will, 2004).
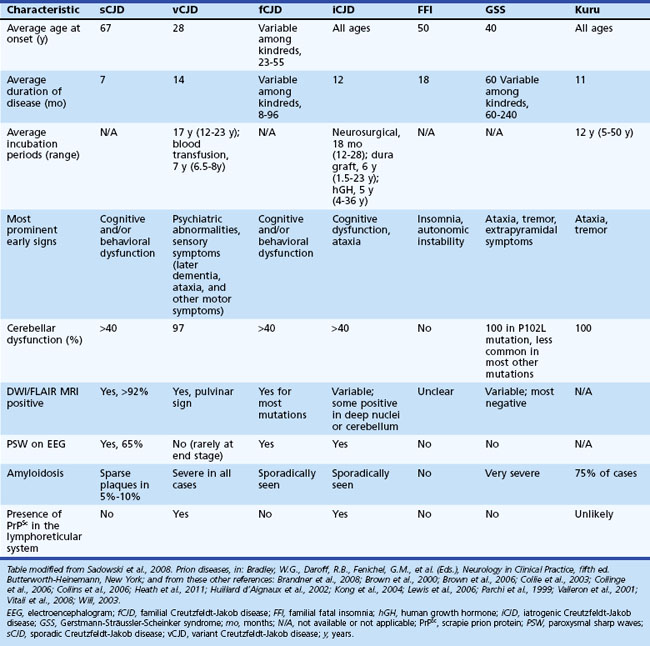
Symptoms of sCJD vary widely but typically include cognitive changes (dementia), behavioral and personality changes, difficulties with movement and coordination, visual symptoms, and constitutional symptoms (Brown et al., 1986a; Rabinovici et al., 2006). Sporadic CJD usually progresses rapidly over weeks to months from the first obvious symptoms to death. The end stage is usually an akinetic-mute state (no purposeful movement and not speaking). Most patients with prion disease die from aspiration pneumonia. Cognitive problems are often among the first symptoms in sCJD and typically include mild confusion, memory loss, and difficulty concentrating, organizing, or planning. Motor manifestations of CJD include extrapyramidal symptoms (bradykinesia, dystonia, tremor), cerebellar symptoms (gait or limb ataxia), and later in the disease, myoclonus (sudden jerking movements). Whereas the cognitive and motor symptoms are often quite obvious, other common early symptoms are often more subtle. These include behavioral or psychiatric symptoms (i.e., irritability, anxiety, depression, or other changes in personality) and constitutional symptoms (i.e., fatigue, malaise, headache, dry cough, lightheadedness, vertigo, etc.). Visual symptoms typically present as blurred or double vision, cortical blindness, or other perceptual problems; they are due to problems with processing of visual information in the brain and not due to retinal or cranial nerve abnormalities. Other symptoms such as aphasia, neglect, or apraxia (inability to do learned movements) due to cortical dysfunction might also occur and can be presenting features. Sensory symptoms such as numbness, tingling, and/or pain are less well-recognized symptoms and are probably underreported, given the magnitude of the other symptoms in sCJD (Geschwind and Jay, 2003; Lomen-Hoerth et al., 2010; Prusiner and Bosque, 2001; Will 2004).
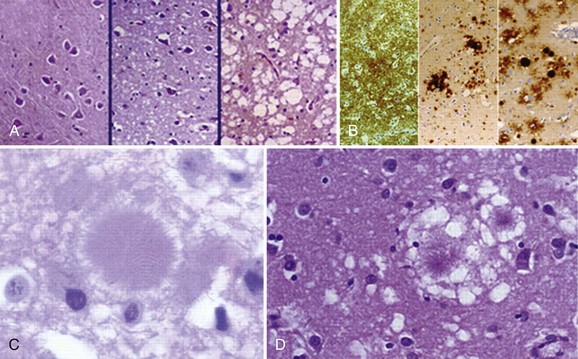
Typical neuropathological features of sCJD include neuronal loss, gliosis (proliferation of astrocytes), vacuolation (i.e., spongiform changes), and deposition of PrPSc (Fig. 53D.3). Animal models suggest the gliosis and vacuolation are early features of prion diseases (Bouzamondo-Bernstein et al., 2004). Some pathologists feel the term vacuolation is the more appropriate term to describe the spongiform changes, as these are not holes but rather fluid-filled vesicles formed in distal dendrites near synapses. Clinical and other features of several prion diseases are shown in Table 53D.1.
Molecular Classification of Sporadic Creutzfeldt-Jakob Disease
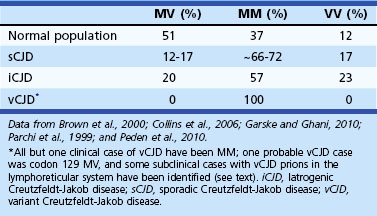
Sporadic CJD has been divided into approximately six molecular subtypes based on the genetic polymorphism at codon 129 in the prion gene (MM, MV, or VV; Table 53D.2) (also see later discussion and Figs. 53D.2 and 53D.6) and the type of protease-resistant prion (type 1 or 2). When PrPSc is extracted from brain and partially digested with proteinase, depending on which of two sites (codon 82 or 97; see Fig. 53D.2) at which PrPSc is cleaved, either a longer 21-kD (type 1) or a shorter 19-kD (type 2) peptide results when run on a Western blot. This classification to some extent separates sCJD cases based on their clinicopathological features. MM1 and MV1 are the most common forms (70%) and present as classic sCJD with rapidly progressive dementia and a duration of just a few months. VV2 (16%) starts with ataxia, later-onset dementia, and a short duration. The remaining four types—MV2 (9%), MM2-thalamic (2%), MM2-cortical (2%), and VV1 (1%)—have a duration of about 1 to 1.5 years. MV2 presents similarly to VV2 with ataxia, but have focal amyloid “kuru” plaques in the cerebellum. MM2-thalamic presents often with insomnia, followed later by ataxia and dementia, with most pathology confined to the thalamus and inferior olives and very little vacuolation; some call this form sporadic fatal insomnia (sFI), as it presents similarly to the genetic prion disease, fatal familial insomnia (FFI). MM2-cortical patients have progressive dementia with large confluent vacuoles in all cortical layers. Sporadic CJD patients with VV1 also present with progressive dementia but have severe cortical and striatal pathology with sparing of the brain stem nuclei and cerebellum. Unlike MM2-cortical, sCJD VV1 patients do not have large confluent vacuoles, but there is faint synaptic PrPSc staining (Parchi et al., 1999). Curiously, as shown in Table 53D.2, heterozygosity at codon 129 in the prion gene, PRNP, is somewhat protective against prion disease.
Diagnosis of Creutzfeldt-Jakob Disease
Several criteria exist for the diagnosis of sCJD. Unfortunately, however, most patients will only fulfill existing criteria at later stages of the disease, because most criteria are designed for epidemiological purposes to ensure that all deceased, non–autopsy-proven cases have a sufficient nonpathological diagnosis. Unfortunately, epidemiological criteria designed for CJD surveillance do not allow diagnosis at early disease stages. Most of these criteria, therefore, are not very helpful when evaluating a patient early in the disease course. Criteria generally categorize patients by level of diagnostic certainty into definite, probable, and possible. Definite criteria require pathological evidence of the presence of PrPSc in brain tissue (by biopsy or autopsy) (Budka 2003; Kretzschmar et al., 1996). Some probable diagnostic criteria are shown in Table 53D.3. The most commonly used probable criteria are the World Health Organization (WHO) Revised Criteria (1998). In the criteria, pyramidal symptoms are motor findings on exam (e.g., hyperreflexia, focal weakness, extensor response). Extrapyramidal symptoms in sCJD typically include rigidity, slowed movement (bradykinesia), tremor, or dystonia, typically due to problems in the basal ganglia or its connections. Akinetic mutism describes when patients are without purposeful movement and mute, and it occurs at the end stage of the disease. Possible CJD criteria are the same as for probable but do not require the ancillary testing (electroencephalogram [EEG] or 14-3-3 tests) (WHO, 1998). Many patients will not meet Revised WHO criteria for probable sCJD until late in the disease course. Criteria utilizing brain magnetic resonance imaging (MRI) were proposed in 2007 (Geschwind, Haman, et al., 2007; Geschwind, Josephs, et al., 2007), and in 2009, Modified European sCJD criteria allowed inclusion of brain MRI (see later discussion). Note that there are errors in these European criteria as published (Zerr et al., 2009), which it is hoped will be corrected in the future (see Table 53D.3). Regardless, there are some potential problems with the European 2009 MRI criteria (see Diagnostic Tests for Sporadic Creutzfeldt-Jakob Disease).
Table 53D.3 Several Diagnostic Criteria for Probable Sporadic Creutzfeldt-Jakob Disease
| WHO 1998 Revised Criteria (WHO, 1998) | UCSF 2007 Criteria (Geschwind et al., 2007) | European Criteria 2009† (Zerr et al., 2009) |
|---|---|---|
CSF, Cerebrospinal fluid; DWI, diffusion-weighted imaging; EEG, electroencephalogram; FLAIR, fluid-attenuated inversion recovery; MRI, magnetic resonance imaging; PSWCs, periodic sharp wave complexes; WHO, World Health Organization.
* Higher focal cortical signs include such findings or symptoms as apraxia, neglect, acalculia, aphasia, etc.
† N.B. There were errors in the table summarizing the criteria in the paper; criteria shown here are derived from the text of the paper.
Diagnostic Tests for Sporadic Creutzfeldt-Jakob Disease
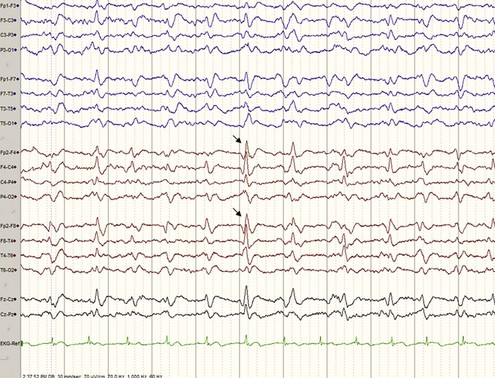
A typical EEG in sCJD has sharp or triphasic waves (periodic sharp wave complexes, or PSWCs) occurring about once every second (Fig. 53D.4); however, this EEG finding is found in only about two-thirds of sCJD patients, typically only after serial EEGs and often not until later stages of the illness (Steinhoff et al., 2004). These EEG findings are relatively specific, but PSWCs are sometimes seen in other conditions including AD, Lewy body disease, toxic-metabolic and anoxic encephalopathies, progressive multifocal leukoencephalopathy, and Hashimoto encephalopathy (Seipelt et al., 1999; Tschampa et al., 2001).
Cerebrospinal fluid (CSF) biomarkers for specific brain proteins have been used for the diagnosis of CJD since 1996, but their clinical utility is somewhat controversial, in part because of varying degrees of sensitivity and specificity around the world. The 14-3-3 protein was one of the first CSF proteins touted as a diagnostic marker for CJD, but its utility is still controversial (Chapman et al., 2000; Geschwind et al., 2003), particularly as it is elevated in many non-prion neurological conditions (Satoh et al., 1999). This test is usually a Western blot that is read subjectively, or at best semiquantitatively, as positive, negative, or inconclusive. Enzyme-linked immunosorbent assay (ELISA) tests were used for a brief period but were found to be unreliable. When first published, the 14-3-3 was reported to have 100% sensitivity and 96% specificity, but this was not a good study because of its small sample size and poor controls (Hsich et al., 1996). Since then, larger European studies have found this protein to have a sensitivity and specificity of about 85%; however, the control patients are not sufficiently characterized in some of these studies (Collins et al., 2006; Sanchez-Juan et al., 2006). Many feel that the 14-3-3 protein is merely a marker of rapid neuronal injury and has poor specificity for sCJD (Chapman et al., 2000; Geschwind et al., 2003; Satoh et al., 1999).
Other potential sCJD CSF biomarkers include total-tau (t-tau), neuron-specific enolase (NSE), and the astrocytic protein, S100β. The sensitivity and specificity of these biomarkers for sCJD varies greatly among studies. One large multicenter European study examined four biomarkers, 14-3-3, t-tau, NSE and S100β; however, not all patients had all four tests done, nor were they necessarily done in the same samples, so this study did not allow legitimate comparison of these biomarkers. Nevertheless, they found the sensitivity and specificity of the 14-3-3 to be 85% and 84%, t-tau (cutoff > 1300 pg/mL) 86% and 88%, NSE 73% and 95%, and S100β 82% and 76%, respectively (Sanchez-Juan et al., 2006). The sensitivity and specificity of these tests in other forms of prion disease such as variant CJD and gPrD are much lower than for sCJD. Some investigators have suggested the ratio of phosphorylated tau (p-tau) to t-tau (p-tau/t-tau) as a good diagnostic test. Additional potential CSF biomarkers, some with a possible functional role in prion diseases, are also being evaluated. Thus far, many feel that t-tau might be the best CSF diagnostic protein for sCJD, but it still is not close to the utility of brain MRI.
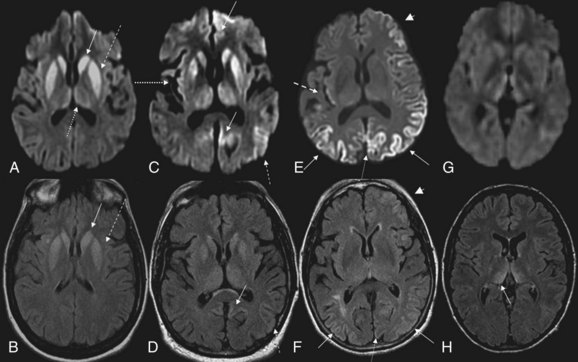
MRI has been shown to be highly sensitive and specific (91%-96%) for sCJD. Although the first MRI abnormalities reported in CJD were basal ganglia hyperintensities, particularly on T2-weighted sequences, cortical gyral hyperintensities, particularly noticeable on fluid-attenuated inversion recovery (FLAIR) and diffusion-weighted imaging (DWI) MRI sequences, are even more common. These cortical hyperintensities are commonly referred to as cortical ribboning. DWI has higher sensitivity than FLAIR. Whenever CJD is suspected, a brain MRI that includes DWI and FLAIR sequences should be obtained (Shiga et al., 2004; Vitali et al., 2008; Young et al., 2005). A typical MRI in sCJD (and some cases of genetic CJD) is shown in Fig. 53D.5. Unfortunately, many radiologists, even at academic centers, are still not familiar with the findings indicative of CJD, and a majority of sCJD MRIs are misread (Wong et al., 2009). Diagnostic criteria for sCJD that included the use of DWI and FLAIR MRI were proposed in 2007 (Geschwind, Haman, et al., 2007; Geschwind, Josephs, et al., 2007), and MRI was included in European criteria in 2009 (Zerr et al., 2009) (see Table 53D.3).
< div class='tao-gold-member'>
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


