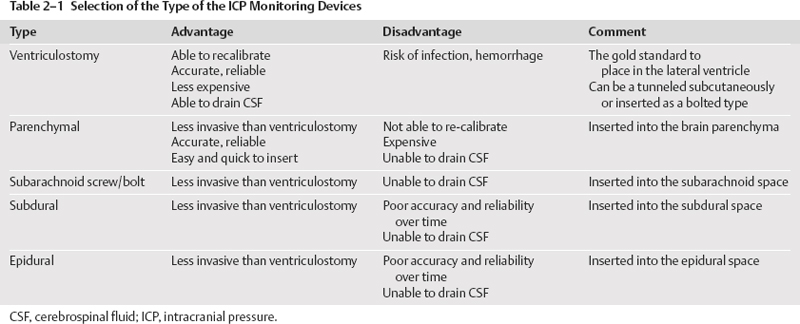
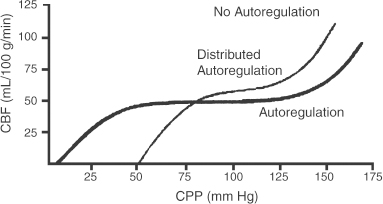
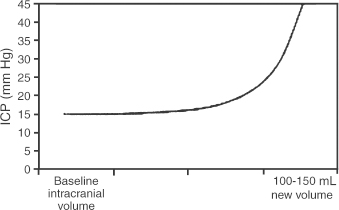
2 Intracranial Pressure Monitoring and Management of Raised Intracranial Pressure Bong-Soo Kim and Jack Jallo One of the most important and common clinical problems encountered by the neurosurgeon is increased intracranial pressure (ICP). During the last several decades, numerous investigations and researches have improved our understanding of the pathophysiology of intracranial hypertension. In addition, the availability of advanced neuroimage and monitoring technologies has resulted in effective management for the patient with central nervous system diseases associated with intracranial hypertension. Uncontrollable intracranial hypertension has been shown to be the primary cause of death in most patients who die of central nervous system diseases such as traumatic brain injury (TBI) and stroke. However, successful management of intracranial hypertension remains a challenge. Virtually no new and effective treatment modality has been identified since ICP monitoring techniques have been available for clinical practice. The goal of this chapter is to discuss up-to-date clinical management of increased ICP. Intracranial hypertension can be a hyperacute emergency requiring reversal if death or profound morbidity is to be avoided. Accurate and real-time ICP monitoring is essential for successful management of increased ICP. Continuous ICP monitoring has several advantages. It identifies increased ICP that may result in a decrease in cerebral perfusion pressure (CPP). Cerebral perfusion pressure is calculated as mean arterial pressure (MAP) minus ICP. In the setting of increased ICP in adult patients, maintenance of CPP above 70 mm Hg is recommended. Intracranial pressure monitoring may provide an early warning of delayed complications. Progressive increase in ICP may indicate the development of intracerebral hemorrhage, cerebral edema, or hydrocephalus. Intracranial pressure data are very useful for predicting the prognosis for recovery after head injury. Intracranial pressure monitoring has been used in the management of patients with brain injuries including TBI, subarachnoid hemorrhage (SAH), intracerebral hematoma, and cerebral ischemia. Decision making with regard to which patients stand to benefit from ICP monitoring can be difficult sometimes. Generally, an ICP monitor should be placed if the condition leading to ICP elevation is amenable to treatment and ICP assessment would be of consequence in decisions for treatment or intervention. Of all clinical indications, ICP monitoring has had the most positive impact on the management of severe traumatic head injury. ICP monitoring can detect changes in pressure before secondary brain injury from ICP occurs. Identifying patients who would benefit from ICP monitoring is based on clinical and radiolographic evaluations. There are unfortunately insufficient data to support a treatment standard for ICP monitoring. The Brain Trauma Foundation guidelines recommend ICP monitoring in patients with severe head injury with an abnormal admission computed tomography scan. Severe head injury is defined as a Glasgow Coma Scale score of 3 to 8 after cardiopulmonary resuscitation. An abnormal computed tomography scan of the head is one that reveals hematomas, contusions, edema, or compressed basal cisterns. In addition, ICP monitoring is appropriate in patients with severe head injury with a normal computed tomography scan if two or more of the following features are noted at admission: age over 40 years, unilateral or bilateral motor posturing, systolic blood pressure <90 mm Hg. Intracranial pressure monitoring is not routinely indicated in patients with mild or moderate head injury.1 However, a physician may choose to monitor ICP in certain conscious patients with traumatic mass lesions. Patients with moderate head injury with contusions of the temporal lobe are an example. The tendency for such injuries to evolve over the first 24 to 48 hours, coupled with their proximity to the brainstem and physical constraint in the temporal fossa, increases the possibility of delayed precipitous deterioration presenting as herniation. Therefore, some institutions tend to monitor such patients using a minimally invasive monitor like intraparenchymal fiber-optic monitor. The primary goal of ICP monitoring is to maintain adequate cerebral perfusion through the use of objective data, and the ICP monitor can be stopped when ICP is in normal range for 24 to 72 hours after withdrawal of ICP therapy. There is no absolute contraindication for ICP monitoring and there are few relative contraindications. Coagulopathy can markedly increase the risk of procedure-related hemorrhage. If possible, the placement of an ICP monitor should be delayed until the international normalized ratio (INR), prothrombin time (PT), and partial thromboplastin time (PTT) are corrected. Generally PT should be less than 13.5 seconds or the INR should be less than 1.4. For emergent situations, fresh-frozen plasma and vitamin K can be given. In our institution, we give a single dose of recombinant coagulation Factor VIIa (Novo Seven) to patients with high INR and PT from Coumadin, who need immediate placement of an ICP monitor or neurosurgical procedures. The platelet count should exceed 100,000/mm3. In patients with a history of taking antiplatelet agents, a pool of platelets should be given, and the platelet function should be assessed with the bleeding time. Immunosuppressants, either iatrogenic or pathologic, are also a relative contraindication for placement of an ICP monitor. There are several methods of classifying ICP monitors. Intracranial pressure monitoring devices are mainly classified according to the location of the monitor and the technology used for determining ICP (Table 2-1). Selection of the type of ICP monitoring device depends on several factors, including the clinical presentation, the need for concomitant cerebrospinal fluid (CSF) drainage, the risks associated with particular devices, system availability, a surgeon’s personal familiarity with such devices, and ease of insertion.2 Increased ICP may be a powerful and independent predictor of outcome in patients with central nervous system disease, particularly severe TBI and stroke. Uncontrollable intracranial hypertension has been shown to be the primary cause of death in nearly half of these patients.3 However, successful treatment of intracranial hypertension remains a challenge. Analysis of ICP data from the Traumatic Coma Data Bank shows that the patients with severe TBI who had hourly ICP >20 mm Hg had significantly worse outcomes.4 Several other clinical studies have shown similar findings. Most neurosurgeons try to maintain ICP below 20 to 25 mm Hg.5,6 CPP represents the pressure gradient acting across the cerebrovascular bed and is a major determinant of cerebral blood flow (CBF). In the presence of intact cerebral autoregulation, CBF remains relatively constant within a wide range of perfusion pressures. This is achieved by vasoconstrictive responses to increased CPP and vasodilatory responses to decreased CPP. Cerebral pressure autoregulation normally has lower and upper CPP limits of ~50 and 150 mm Hg, respectively. (Figs. 2-1 and 2-2) When CPP is outside the bounds of pressure autoregulation, CBF becomes directly dependent on CPP. In such situations, CPP must be maintained above a critical minimum to prevent cerebral hypoperfusion and below a critical maximum to prevent hyperemia, vasogenic edema, and increased ICP. Figure 2-1 Cerebral autoregulatory curves. (From Marmarou A. Physiology of the Cerebrospinal Fluid and Intracranial Pressure. In: Winn RH (ed). Youman’s Neurological Surgery, 5th ed. Philadelphia: Elsevier; 2004: 181–183.) Reprinted by permission. CBF, cerebral blood flow. The traditional practice of elevating the head at 30 to 45 degrees above the heart to lower ICP in head-injured patients has been challenged in recent years. Some argue that patients with intracranial hypertension should be placed in a horizontal position, which maximizes CPP and reduces the severity and frequency of pressure-wave occurrence. However, ICP is generally significantly higher when the patient is in the horizontal position.7 Recent data indicate that head elevation to 30 degrees significantly reduces ICP without reducing CPP or CBF. The neck must be maintained in a neutral position and compression of the jugular veins must be avoided so as not to compromise jugular venous outflow. The onset of action of head elevation is immediate. Because agitation, anxiety, pain, and uncontrolled movement can contribute to undesirable increases in ICP and cerebral metabolic demands, the use of sedatives and pharmacological paralytics can play an effective role in managing increased ICP, especially in severe head injury. However, such drugs can alter the neurological examination and must be used with prudence. There is no real preference for one sedative over another; the key factor is that hypotension secondary to excessive doses of a sedative should be avoided and is more prone to occur in patients with underlying hypovolemia. Additionally, shorter acting agents allow for intermittent clinical examination. Propofol is being increasingly used for patients in the neurosurgical intensive care unit (ICU), particularly for head-injured patients. Propofol is potentially advantageous in this setting, given its wide dose response, short elimination half-life (24 to 64 minutes), potent anticonvulsant and neuroprotective effects. In contrast with the benzodiazepines and opiates, long-term propofol use does not result in addiction or withdrawal phenomena.8 Increasing dosage requirements, however, may occur. Whether this problem is related to tolerance or an increased rate of drug clearance remains unclear. Propofol causes hypotension, particularly in volume-depleted patients. The tendency toward hypotension with propofol can be minimized if patients have a normal intravascular volume before initiating propofol; the infusion begins at a rate of less than 20 mcg/kg/minute and does not increase by more than 10 mcg/kg/minute every 5 minutes.9 Prolonged use (>48 hours) of high doses of propofol (>66 mcg/Kg/min) has been associated with lactic acidosis, bradycardia, and lipidemia in pediatric patients. A rare complication first reported in pediatric patients and also observed in adults is known as “propofol syndrome,” characterized by myocardial failure, metabolic acidosis, and rhabdomyolysis. Hyperkalemia and renal failure have also been associated with this syndrome. Hypertriglyceridemia and pancreatitis are uncommon complications.10 Morphine, fentanyl, and sufentanil are common analgesics for sedation in the intensive care unit and do not change ICP.11 Etomidate is used to facilitate endotracheal intubations; however, even a single bolus of etomidate can cause relative adrenal insufficiency in patients with TBI. Etomidate should be avoided.12 Midazolam can be used alone or in combination with morphine infusion. Care must be taken to avoid hypotension. Midazolam can be reversed with flumazenil when needed; however, uncontrolled ICP could result from overly rapid reversal. Although pharmacological paralysis decreases ICP in patients with refractory intracranial hypertension, early, routine, long-term use of neuromuscular blocking agents in patients with severe head injuries to manage ICP does not improve overall outcome and may actually be detrimental because of the prolongation of their ICU stay and the increased frequency of extracranial complications, such as pneumonia and respiratory failure, associated with pharmacologic paralysis.13 Figure 2-2 Intracranial pressure volume curve. (From Marion DW. Pathophysiology and Treatment of Intracranial Hypertension. In: Andrew BT (ed). Intensive Care in Neurosurgery. New York: Thieme Medical Publishers; 2003; 47.) Reprinted by permission. ICP, intracranial pressure. Figure 2-3 Algorithm for elevated intracranial pressure. GCS, Glasgow Coma Scale; ICP, intracranial pressure; CPP, cerebral perfusion pressure. Osmotic diuretics have been widely used in the treatment of increased ICP. Although no Class I evidence has been reported for comparing the efficacy of either mannitol or hypertonic saline, Class II and III evidence suggests that both agents may be effective in reducing ICP. However, many different regimens (concentration, dose, bolus versus continuous infusions, and duration) have been utilized. Unfortunately, comparisons between these treatment protocols are lacking.14 The most commonly used diuretic is mannitol. The ICP-reducing effects of mannitol are likely to depend on several mechanisms, including an osmotic effect, a diuretic effect, and a hemodynamic effect. Traditionally, the effect of mannitol on ICP was attributed to ”brain shrinkage” resulting from pulling water from the brain’s interstitial space into the intravascular compartment. This effect depends on the establishment of osmotic gradients between plasma and cells. Gradients as low as 10 mosmol/L seem to be effective in reducing ICP. It may take up to 30 minutes to develop these patients.15 For mannitol to be effective, the blood-brain barrier (BBB) must be preserved. Thus, an impaired BBB theoretically limits the efficacy of osmotic diuretics because an osmotic gradient cannot be formed. An osmotic gradient must be formed to drive water from the brain into the intravascular compartment. However, mannitol almost always reduces increased ICP, regardless of its cause. When 1 g mannitol/kg body weight is given over 10 minutes, a rise in serum osmolarity of 20 to 30 mosmol/L occurs and returns to the control level in ~3 hours.16 (The diuretic effect of mannitol may also contribute to ICP reduction.14) The direct removal of water from brain parenchyma is only partially responsible for the observed ICP reduction that follows mannitol administration. Following a bolus infusion of hyperosmolar mannitol, water is drawn from the tissues, including red blood cells, to the plasma. This immediate plasma-expanding effect reduces blood viscosity by decreasing the volume, rigidity, and cohesiveness of red blood cells.17 Altered blood rheology results in reduced cerebrovascular resistance, increased CBF, and increased CPP. Autoregulatory vasoconstriction may then decrease cerebral blood volume (CBV) and ICP. These immediate rheological effects of mannitol may be the primary mediators of ICP reduction.18 Mannitol is known to open the BBB, possibly by dehydrating endothelial cells and thus causing separation of tight junctions.19 If endothelial cells are swollen in an area of brain edema, mannitol may be of benefit in increasing CBF via increasing capillary inner diameter by decreasing the endothelial cell swelling. Mannitol may produce immediate hypotension after rapid infusions, especially in volume-depleted patients. Renal failure is one of the most important side effects. Significant concern over renal failure often limits the use of mannitol. Although not well understood, possible mechanisms for mannitol-induced renal failure include renal afferent arteriolar vasoconstriction, renal tubular swelling, tubular vacuolation, increased intraluminal sodium concentration at the level of macula densa, and elevation of plasma oncotic pressure.20–22 Traditional treatment guidelines in clinical practice recommend that mannitol should not be administered if serum osmolarity level exceeds 320 mosmol/L because of concern about inducing renal failure.23–25 However, Gondim et al recently reported no relationship between osmolarity and renal insufficiency. Patients with preexisting conditions that are likely to impair renal function chronically appear to be at higher risk.26 Intermittent boluses of mannitol (0.25 to 1 g/kg body weight) are recommended over continuous infusion because continuous infusion is more likely to cause rebound increased ICP, particularly in cases of prolonged use of mannitol with rapid discontinuation. There are several theoretical mechanisms to explain rebound phenomenon. The most widely held explanation is due to penetration of osmotically active solutes into the edematous brain, their accumulation creating an unfavorable reversal of the osmotic gradient. Marshall et al have shown that reduction of ICP with a dose of 0.25 g/kg is equal to the response obtained with doses of 0.5 to 1.0 g/kg.27 Hypertonic saline (HTS) has proven useful in the control of elevated ICP, especially when other treatments have failed. Suarez described eight patients (one TBI, several subarachnoid hemorrhage (SAH), one glioma) administered 30-mL boluses of 23.4% HTS when refractory to mannitol.28 All showed lowering of their ICP from a mean of 41.5 to 17 mm Hg for several hours. There was no increased serum Na+ despite multiple doses, but no change in central venous pressure (CVP) or urine output either. In another report, nine patients with cerebral vascular accident (CVA) received either 7.5% HTS or mannitol.29 Between these patients, 30 instances of increased ICP or dilated pupils were randomly treated with either agent. An improved ICP (lowered ≥10%) or resolution of pupil abnormality was found in 10/14 patients given mannitol, and all the HTS patients. A greater absolute reduction and a faster response were noted with HTS. However, the CPP improvement was better with mannitol. Retrospective studies in children have also produced positive results. A study of 68 patients with 3% HTS therapy for refractory intracranial hypertension had good control.30 They reported only three deaths from refractory intracranial hypertension, which was less than expected for the severity of injury. This study, however, did not include patients with “nonsurvivable injuries.”31 Gemma et al described one patient with vertebral spasm and ischemic brainstem injury after TBI.32 The patient received 2.7% and 5.4% HTS for 48 hrs each, with somatosensory evoked potentials (SSEPs) and the neurologic exam showing sustained improvement from 24 hours after initiation of therapy. Similar to mannitol and other osmotic agents, its HTS’s ICP-reducing effects are dependent on several mechanisms. Hypertonic saline infusion increases the osmotic gradient between the brain and the blood, and draws fluid from the interstitial space to the intravascular space.33–38 Cerebral edema can be caused by leakage from damaged microvasculature (BBB dysfunction), vasoregulatory dysfunction, and accumulation of osmotic molecules in the interstitial and the intracellular space of the ischemic brain. Cell death and lysis release osmolytes into the interstitial space. Ischemic cells in the penumbra, unable to complete the metabolic cycle, collect metabolic products in the intracellular space, resulting in higher than normal parenchymal osmolarity in the entire region of the injured brain.39–41 The increased serum osmolality with HTS infusion reduces the perceived osmotic gap, and also reduces CSF production, which can improve intracranial compliance. Human trials showed ICP improvement for ~72 hours when Na+ levels increased 10 to 15 mEq/L with HTS therapy.42,43 Hypertonic saline, as both bolus and continuous infusions, lowers ICP.34–38,43 There is no evidence yet supporting one concentration of HTS above others in efficacy for controlling cerebral edema. Some studies show this effect wanes, and ICP increased to baseline levels when isotonic fluids were used for maintenance after an initial HTS bolus.44,45 Even with prolonged hypernatremia, tolerance to HTS develops after several days.43,45 The mechanism appears to be movement of cerebral osmolytes by active transport into cells in response to TBI, with increased intracellular osmolarity and loss of the osmotic gradient.45 These osmolytes are organic molecules including some amino acids (glutamate, glutamine, g-aminobutyric acid, N-acetylaspartate, alanine, aspartate, and taurine), polyhydric alcohols (myoinositol), and methyl amines (creatine and glycerophosphorylcholine).46,47 This process occurs after 3 days of a maintained hypertonic state. Sustained hyperosmolarity also increases vasopressin release and thirst, due to osmoreceptors in periventricular regions such as the lamina terminalis, which projects to the hypothalamus.48,49 Increases in the MAP by a hemodynamic effect of HTS infusion have been documented in human models of cardiogenic, septic, and hemorrhagic shock.50–56 This has been shown to be due to multiple additive effects. HTS increases intravascular volume by causing fluid to enter the intravascular compartment.44 It may also increase cardiac output by hormonal action.57 The benefits of a higher MAP are accompanied by prevention of fluid-overloading and hemodilution because much smaller volumes are needed. The beneficial effect on MAP is temporary (15 to 75 minutes), but can be extended by the addition of colloid.32 This is likely because the intravascular volume remains higher for a longer period of time, for Na+ and Cl– can cross the capillary endothelial membranes in the rest of the body, and draw intravascular fluid into the interstitial space, whereas colloids remain in the intravascular space. Hypertonic saline therapy also has a vasoregulatory effect. Cerebral ischemia precipitated by vasomotor dysfunction is one cause of secondary brain injury.58–60 Studies have also documented ischemia due to cerebral edema and vasospasm, as well as hyperperfusion in the first 2 weeks after injury.61–63 Hypertonic saline therapy increases capillary vessel inner diameter and plasma volume, which counteracts vasospasm and hypoperfusion by increasing CBF. This action may come about through dehydration of endothelial cells and erythrocytes, increasing the internal diameter of vessels and improving movement of red blood cells through cerebral capillaries.64 Hypertonic saline therapy simultaneously prevents increased ICP from hyperperfusion.65 The net effect increases cerebral oxygen delivery and improves PaO2 by improved CBF and decreased pulmonary edema.66 Primary brain injury during trauma causes extensive neuronal depolarization, increasing extracellular glutamate. Then, secondary ischemia reduces the amount of ATP production, which prevents the homeostatic function of active transport transmembrane Na+/K+ exchange pumps.67–72 The resulting lower extracellular Na+ reverses the direction of the Na+/glutamate passive cotransporter, increasing extracellular glutamate. Increased phospholipase activity and increased membrane permeability allow leakage of additional glutamate from the cell. The higher intracellular sodium concentration also causes diffusion binds to cell surface receptors and opens Ca2+ channels, increasing the diffusion of water into the cell, opening stretch-sensitive channels that allow further release of glutamate. This leads to a positive feedback loop and can cause massive cell death.73 Hypertonic saline can prevent pathologic glutamate release, since increased extracellular Na+ returns the Na+/ glutamate pump to its normal function of glutamate reuptake. The intracellular concentrations of Na+, Cl–, and resting membrane potential are also restored. The Na+/Ca2+ pump is activated to reduce intracellular Ca2+, thereby limiting neuronal excitation.73 Hypertonic saline therapy has multiple immunomodulatory effects. Alterations in prostaglandin production and increases in cortisol and ACTH levels have been noted.74 It has also been shown to decrease leukocyte adherence and migration.75 Despite suppressive effects on the inflammatory system, infusion of HTS reduces the rate of infectious complications.76 It reduces CD4+ suppression, and normalizesNK cell activity in rat models. Hypertonic saline infusion in hemorrhagic shock models also limits the amount of bacterial translocation, reducing the risk of bacterial seeding and sepsis. Thus, HTS acts through multiple parallel complementary and interacting pathways to produce complex effects on multiple systems. The net effect is to reduce ICP and improve cardiovascular function to reduce secondary brain injury and thus, it is hoped, improve outcomes. Hypertonic saline therapy is not without potentially adverse effects. The most serious theoretical complication of HTS therapy is the development of central pontine myelinolysis (CPM). This is the destruction of myelinated fibers after a rapid rise in serum sodium, most commonly affecting deep white matter, the pons being most susceptible. Cerebral osmolytes play a significant role in CPM, as their concentration and diffusibility affect the osmolality.77 Literature from prospective animal studies and human case reports of correction of hyponatremia recommend increasing Na+ no more than 10 to 20 mEq/L/day.78 However, human trials with HTS have not documented very rapid increases in Na+, with no reported cases of CPM either.28,30,42 The use of HTS has led to documented cases of renal insufficiency and even failure, although it is less common than with the use of other osmotic diuretics used to control cerebral edema. A quadrupled rate of renal failure in burn patients receiving HTS for resuscitation versus lactated ringer’s solution (LR) was noted, but data from this patient population, with large fluid losses, may not apply to TBI cases.79 In 2 of 10 pediatric TBI patients with continuous HTS maintenance fluids, temporary renal insufficiency was noted, which occurred after the peak Na+ had already been passed, and was temporally associated with septic episodes. The renal failure, therefore, may not have been due to osmotic effects, but to hypotension.43 Hemorrhage secondary to excessive fluid resuscitation has been reported with HTS.66,80 This is usually associated with uncontrolled primary hemorrhage. One proposed explanation for the observed coagulopathy is the dilution of plasma constituents with rapid intravascular volume expansion.81 Decreased platelet aggregation with increased PT/PTT with ≥10% plasma replacement has also been observed.82 Rapid plasma volume expansion by HTS can be associated with fluid overload, particularly in patients with a preexisting heart failure. No cases of congestive heart failure or pulmonary edema were found in a retrospective study of 29 patients with SAH and hyponatremia, on continuous 3% HTS infusions.28 Hypokalemia and hyperchloremic acidosis have both been observed when no K+ or acetate replacement was used concurrently with HTS administration.28,38,42 These abnormalities are easily prevented by prophylactic administration of KCl, and using HTS solutions with 50/50 Cl–/ acetate. Although HTS has been quite effective in reducing high ICPs, a rebound increase has been reported with bolus HTS doses, or after continuous HTS infusions were stopped, or even after 24 hours of continuing HTS infusion in TBI patients.38,42,43 This may be due to the intrinsic half-life of HTS effects. However, compared with mannitol, HTS is less likely to cross the BBB and therefore less likely to cause rebound cerebral edema.83 Hyperventilation has been used in the management of intracranial hypertension for decades since Lundberg et al reported its use to lower increased ICP.84 The reactivity of the cerebral vasculature to carbon dioxide (CO2) is one of the primary mechanisms involved in the regulation of CBF.85,86 Carbon dioxide reactivity involves smaller pial arteriole, and larger intracranial vessels are not significantly affected by changes in PaCO2.87,88 In vivo, very localized perivascular changes of PaCO2 or pH can change the vascular diameter, indicating that elements of the vascular wall are responsible for effecting changes in the diameter of blood vessels. The vascular endothelium, smooth muscle cells, and extravascular cells (the perivascular nerve cells, neurons, and glia) may be involved. Changes in pH may exert their effect on smooth muscle tone through second messenger systems or by altering the calcium concentration in vascular smooth muscles directly. Various agents have been identified as potential second messengers, including prostanoids, nitric oxide (NO), cyclic nucleotides, potassium, and calcium.89 Cerebral blood flow changes by approximately 3% for each mm Hg change in PaCO2 over the range of 20 to 60 mm Hg. 90,91 The relationship between PaCO2 and ICP is not linear and the greatest effect is between PaCO2 values of 30 and 50 mm Hg in humans.92 Carbon dioxide reactivity is preserved in most patients with severe head injury,90,93 so hyperventilation can rapidly lower ICP through the reduction in CBV in patients with severe head injury. A recent study demonstrated that a blood volume change of only 0.5 mL was necessary to produce an ICP change of 1 mm Hg in patients with severe head injury.94 A lower blood volume was necessary to produce a significant ICP change in patients with reduced compliance. Further, it was shown that the effects on ICP were greater during hypercapnia than during hypocapnia. In spite of the wide use of hyperventilation in the treatment of intracranial hypertension, several studies illustrate deleterious effects of hyperventilation on CBF, cerebral oxygenation, and metabolism. Using positron emission tomography imaging in the patients with severe head injury Cole’s demonstrated that even mild hyperventilation (PaCO2 <34 mm Hg) could reduce global CBF and increase the volume of critically hypoperfused brain tissue, despite improvements in CPP and ICP.95 Surprisingly, only a few studies have addressed the important question of whether beneficialeffects on ICP remain present during prolonged hyperventilation. Only one prospective randomized clinical trial has been reported concerning the effect of hyperventilation on clinical outcome. Muizelaar et al compared the outcomes of patients who were prophylactically hyperventilated to a PaCO2 of 25 mm Hg for 5 days to patients in whom the PaCO2 was kept at 35 mm Hg. At both 3 and 6 months after injury, patients with an initial Glasgow Coma Scale motor score of 4 or 5 had a significantly better outcome when they were not hyperventilated.96,97 Brain Trauma Foundation Guidelines recommend that prophylactic hyperventilation (PaCO2 ≤35 mm Hg) therapy during the first 24 hours after severe TBI should not be used because it can compromise cerebral perfusion during a time when CBF is reduced. In the absence of increased ICP, chronic prolonged hyperventilation therapy (PaCO2 <25 mm Hg) should be avoided after severe traumatic brain injury. Jugular venous oxygen saturation (SjO2), arterial jugular venous oxygen (AVdO2) content differences, brain tissue oxygen monitoring, and CBF monitoring may help to identify cerebral ischemia if moderate hyperventilation (PaCO2 <30 mm Hg) is necessary.1,98 Acute hyperventilation has an established role in the emergency management of acute neurological deterioration -when there are clinical signs of herniation or acute, severe elevations in ICP. Hyperventilation of brief duration may then be life-saving until definitive management can be undertaken.1,85,98 It has long been known that barbiturates can reduce ICP in a variety of clinical conditions that are associated with brain swelling.99–101 The precise mechanism of ICP reduction by barbiturates is not clearly defined. But, one is thought to be hemodynamic alternation because of the immediate effect on ICP. Barbiturates cause a dose-dependent reversible depression of neuronal activity associated with a reduction in the cerebral metabolic rate, and it is thought that autoregulatory flow-metabolism coupling then results in reductions in CBF and CBV, thereby causing a decrease in ICP.102,103 Barbiturates also alter cerebrovascular tone.102,104 In addition, barbiturates act as free radical scavengers and may limit peroxidative damage to lipid membranes.105–107 Substantial side effects and complications of barbiturate therapy have been reported in the literature. They may occur despite thorough and appropriate clinical monitoring.108,109 The most common and important complication is arterial hypotension from myocardial depression and decreased systemic vascular resistance. Hypotension caused by barbiturates is treated first with volume replacement and then with vasopressors such as dopamine or neosynephrine, if necessary. Laboratory studies suggest that for the treatment of hypotension associated with barbiturate coma, volume resuscitation may be better than vasopressors.110 Other complications during the treatment of intracranial hypertension with barbiturate coma include hypokalemia, respiratory complications, infectious hepatic dysfunction, renal dysfunction, and hypothermia.111–113 Indications for initiating barbiturate therapy have not been clearly defined. Because of the critical hypotension associated with barbiturates, and because a neurological examination cannot be performed during treatment, barbiturate coma is usually reserved for patients with intracranial hypertension resistant to other modalities. Thus chances for a favorable outcome are greatest in younger patients without evidence of brainstem injury and without significant hemodynamic instability. Pentobarbital and thiopental are relatively short-acting barbiturates. Thiopental is administered as a loading dose of 5 to 10 mg/kg, followed by a continuous infusion of 3 to 5 mg/kg/hr. Pentobarbital is also given in both loading and maintenance doses. The loading dose is 10 mg/kg, given over 30 minutes, followed by 5 mg/kg each hour for three doses. This typically provides a therapeutic level after the fourth dose. The maintenance dose is 1 to 3 mg/kg per hour, adjusted so that either the serum level is in the therapeutic range of 30 to 50 mcg/mL or the electroencephalogram has a burst suppression pattern. Winer et al showed that plasma and CSF pentobarbital levels do not accurately reflect the physiologic effects of pentobarbital, and recommended monitoring the electroencephalogram instead of pentobarbital levels.114 However, if the electroencephalogram is not available immediately, initiating barbiturate therapy should not be delayed. When barbiturate therapy is undertaken, continuous monitoring of all physiologic parameters is critical. Therefore, a Swan-Ganz catheter is placed to monitor directly cardiac output, pulmonary wedge pressure, and peripheral vascular resistance in all patients. Although high doses of barbiturate therapy have been used to treat increased ICP widely since the 1970s and several clinical trials were conducted,5,99,113,115 none of these studies clearly proved the efficacy of barbiturates. Ward et al could not show any superiority of prophylactic barbiturate coma in a randomized trial of severely head- injured patients.113 Schwab et al found that barbiturate coma in the therapy of increased ICP after severe hemispheric stroke can provide a short-term reduction of elevated ICP, like other conservative measures of ICP control such as osmotic therapy and hyperventilation, but that it fails to achieve sustained ICP control.115 The most comprehensive review and meta-analysis showed that there was no evidence that barbiturate therapy in patients with acute severe head injury improved outcome. However, a randomized multicenter trial demonstrated that instituting barbiturate coma in patients with refractory ICP resulted in a 4-fold greater chance of controlling ICP.5 The beneficial effects of mild to moderate hypothermia in experimental models of TBI have been demonstrated in a large number of laboratory studies.116–119 The mechanism by which hypothermia may offer neuroprotection is not clearly defined, but is thought to be multifactorial. Hypothermia can decrease the cerebral metabolic rate.120 Autoregulatory reductions in CBF and CBV may then decrease ICP. Experimental studies in animal models have demonstrated that mild or moderate hypothermia decreases cerebral edema, reduces BBB dysfunction, and reduces extracellular levels of excitatory neurotransmitters and free radical production.106,121,122 Adverse effects of hypothermia include a higher incidence of cardiac arrhythmias, coagulopathies, a decrease in platelet count, pulmonary infections, hypothermia-induced diuresis, pancreatitis with high serum amylase and lipase, and electrolyte derangements. Therefore, hypothermia was applied using a strict protocol to prevent the occurrence of side effects.117,123–127 Several promising experimental trials and case series have suggested that induced hypothermia could be of benefit in reducing the risk of both the poor neurologic outcome and death in patients with severe TBI.55,117,119,128–130 However, several clinical trials including multicenter randomized trials have reported the efficacy of induced hypothermia in decreasing the mortality and morbidity associated with severe TBI with conflicting results.117,118,131–133 McIntyre et al recently reviewed and analyzed 12 randomized controlled trials of therapeutic hypothermia. They found that therapeutic hypothermia was associated with a 19% reduction in the risk of death and a 22% reduction in the risk of poor neurologic outcome, compared with normothermia. Hypothermia longer than 48 hours was associated with a reduction in the risks of death and of poor neurologic outcome, compared with normothermia. Hypothermia to a target temperature between 32°C and 33°C, a duration of 24 hours, and re-warming within 24 hours were all associated with reduced risks of poor neurologic outcome, compared with normothermia.132 However, it should be noted that the meta-analysis does not indicate that mortality is lowered in groups of patients with TBI as a result of induced hypothermia as conducted in the majority of these studies. Thus, any conclusions regarding the use of hypothermia in head-injured patients are controversial and not strongly indicated by the current level of evidence.131 Animal studies have shown that hypothermia can alter many of the damaging effects of cerebral ischemia. Intraischemic hypothermia reduced infarct size in most occlusion models. Tissue salvage with delayed onset of hypothermia was less dramatic, but was commonly observed, when hypothermia was begun within 60 minutes of stroke onset in permanent and 180 minutes of stroke onset in temporary occlusion models. Prolonged postischemic hypothermia further enhances efficacy. Studies have shown that intraischemic hypothermia is more protective than postischemic hypothermia, and more benefit is conferred in temporary than in permanent occlusion models. The efficacy of postischemic hypothermia depends on the time of initiation and the duration and depth of hypothermia.134 Although hypothermia is remarkably neuroprotective in animal models, it may lack efficacy in human trials because it may be underdosed or overdosed. Adverse systemic effects may outweigh the benefits of brain hypothermia in a clinical trial. An open pilot study on the efficacy of induced moderate hypothermia showed hypothermia could improve clinical outcome in malignant middle cerebral artery (MCA) infarction. Authors reported 44% of the mortality rate with moderate hypothermia, compared to a mortality rate of ~80% with standard treatment.135,136 Glucocorticoids have been valuable adjuncts in the management of patients with intracranial tumors, both primary and metastatic. Focal neurologic deficit and decreased mental status due to peritumoral vasogenic edema may improve within hours of surgery.137 The exact mechanism of the action of steroids remains unclear. The most common regimen is dexamethasone, but methylprednisolone can be substituted. The vasogenic edema from brain abscess may be improved with the steroid. However, the therapeutic usefulness of steroids for abscess is controversial. Some authors believed that reducing peri-abscess inflammation with steroids may worsen outcome by decreasing delivery of antibiotics to the infected area.138 Therefore, many authors recommend that steroids be reserved for cases in which mass effect is causing life-threatening herniation.139,140 It is clear that steroids decrease the frequency of deafness and other neurologic deficits in children. Corticosteroids are now the standard of care in pediatric patients with meningitis. However, it is important to note that mortality has not been changed in studies to date.109 In most other situations involving increased ICP such as TBI, ischemic stroke, hemorrhage, and hypoxic encephalopathies, the routine use of steroids has not been shown to be beneficial and may be harmful.99,141,142 Drainage of CSF is the most effective and rapid way of decreasing ICP. Drainage of a small amount CSF can be very effective in lowering ICP. A ventricular catheter provides for measurement of ICP and also CSF drainage for treatment of increased ICP. Because it requires penetration of the brain parenchyma in patients who often have coagulopathy, there is the risk of a ventriculostomy-related hematoma. Risk of significant hematoma requiring surgical evacuation is ~0.5%.6 Infection is another important ventriculostomy-related complication. Risk factors for ventriculostomy-related infections include intracerebral hemorrhage with intraventricular hemorrhage, neurosurgical operations including operation for depressed skull fracture, ICP of 20 mm Hg or more, ventricular catheterization for more than five days, and irrigation of the system. Although there is no consensus regarding the use of prophylactic antibiotics with ICP monitors and ventriculostomy, most institutions use prophylactic antibiotics for ventriculostomy. Other complications of ventriculostomy include failure of optimal placement, malfunction or obstruction of drainage, and seizure. If ICP is elevated because of a space-occupying lesion, medical intervention alone may not satisfactorily normalize ICP. Patients frequently benefit from removal of the intracranial lesion. Traumatic head injury patients with intracranial hematomas are frequently surgical candidates, depending hematoma size, location, mass effect, or clinical condition, especially if the hematoma is epidural or subdural (Table 2-2). Surgical evacuation of spontaneous intracerebral hemorrhage remains controversial unless used as a lifesaving measure. The majority of spontaneous intracerebral hemorrhage is seated deep in the basal ganglia and thalamus and is related to hypertension. Several clinical studies have shown that no evidence of better clinical outcome was found in surgical evacuation over the best medical treatment for treatment of deep-seated intracerebral hemorrhages.143,144 However, certain factors should be considered when evaluating surgical candidacy of patients with spontaneous intracerebral hemorrhage. Patients with significant mass effect and impending herniation may benefit from emergent surgical evacuation. However, comatose patients with evidence of lost upper brainstem reflexes and extensor posturing do poorly, regardless of surgical intervention.145 Certainly hemorrhage in the cerebellum benefits from evacuation, especially if there are signs of obstructive hydrocephalus or compression of the brainstem, or the size of the hematoma is more than 3 cm in diameter. For patients with a brain tumor, decision making for surgical resection is complex unless herniation is impending. Several factors including number, size, and location of lesions, as well as expected response of the tumor type to radiotherapy and chemotherapy, should be considered. Despite the lack of prospective, randomized, controlled trials to define the role of decompressive craniectomy, the value of decompressive craniectomy for increased ICP associated with several neurosurgical conditions has been well reported. Disappointing experience in the past with decompressive craniectomy and lack of Cass I evidence, the data from recent studies on decompressive craniectomy for refractory intracranial hypertension have indicated an improved outcome compared with outcome following medical management.146–152 Most patients with an MCA stroke experience unilateral brain swelling and brain distortion which may lead to transtentorial herniation, which has up to an 80% mortality rate.7,153,154 Recent literature reviews conclude that a significant mortality rate reduction (16 to 40% reduction in mortality rate), a wide therapeutic window (2 to 3 days), and a low incidence of intraoperative complications make decompressive craniectomy a relevant treatment in malignant MCA infarction.155 Gupta et al reported in their review of 12 clinical series that age may be a crucial factor in predicting functional outcome after hemicraniectomy for large MCA territory infarction.155 Good functional outcome following early emergency craniectomy for hemorrhagic infarct secondary to venous sinus thrombosis was reported, despite fixed and dilated pupils prior to operation.156 A recent paper also describes good to excellent outcome following unilateral hemicraniectomy for poor grade SAH patients with a large Sylvian fissure hematoma.106 Aarabi et al found that in patients with severe head injury and brain swelling, those with an admission Glasgow Coma Scale (GCS) score greater than 6 are especially good candidates for decompressive craniectomy.146 The European Brain Injury Consortium (EBIC) and the joint Brain Trauma Foundation (BTF) and American Association of Neurological Surgeons (AANS) guidelines for severe head injuries describe decompressive craniectomy as a therapeutic option for brain edema that does not respond to conventional therapeutic measures.98,157 Although successful management of intracranial hypertension remains a challenge and virtually no new and effective treatment modality has been identified, numerous clinical studies have investigated the effectiveness of management modalities of intracranial hypertension. The ineffectiveness of traditional long-standing management practices like steroids, anticonvulsants for preventing late seizures, and chronic hyperventilation was highlighted for management of the patient with severe TBI. Although recommendations for the management of patients with intracranial hypertension are entirely based on Class II and Class III evidence, treatment guidelines and protocol-driven therapy for the management of patients with intracranial hypertension have increased favorable outcomes when compared with historical controls. This improvement of outcomes is the result of more rational and scientifically justified application of standard practice protocols such as the guidelines for severe TBI from the Brain Trauma Foundation.
Intracranial Pressure Monitoring
Indications for Intracranial Pressure Monitoring
Contraindications for Intracranial Pressure Monitoring
Types and Selection of Intracranial Pressure Monitors
Management of Raised Intracranial Pressure
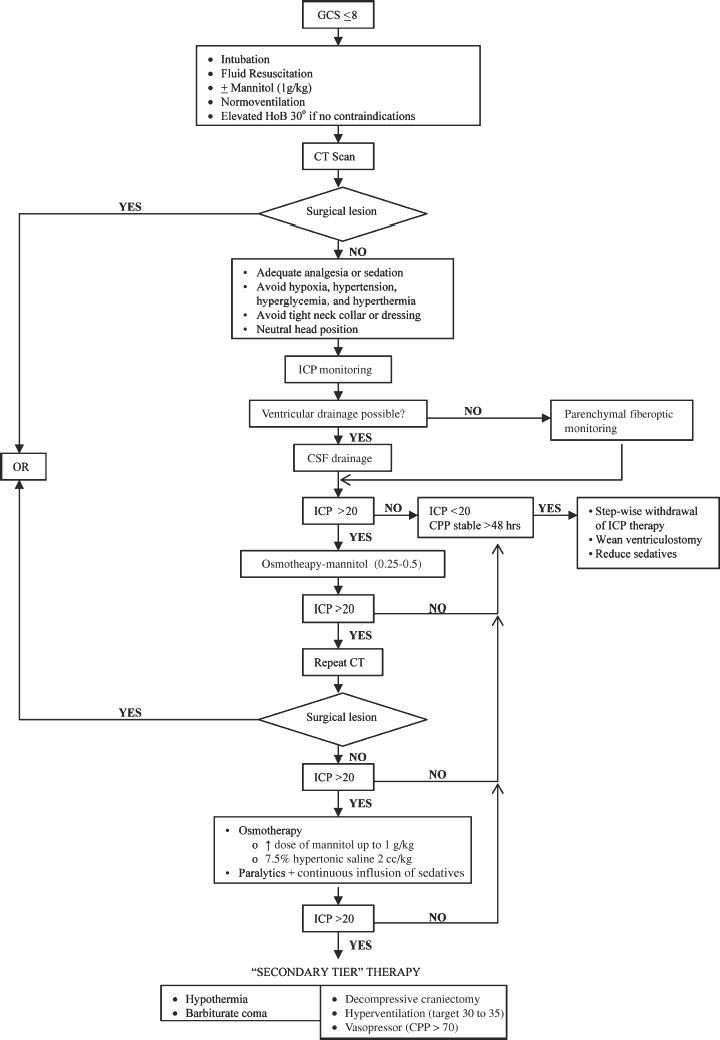
Medical Treatment of Raised Intracranial Pressure (Fig. 2-3)
Head Position
Sedatives and Paralytics
Osmotic Therapy
Mannitol
Hypertonic Solutes
Hyperventilation
Barbiturates
Hypothermia
Steroids
Surgical Treatment of Raised Intracranial Pressure
CSF Drainage
Resection of Source of Mass Effect
Decompressive Craniectomy
Conclusion
| Type of Lesion | Indications for Surgery | Timing |
| Acute epidural hematoma | * An EDH greater than 30 cm3 should be surgically evacuated regardless of the patient’s GCS score. | It is strongly recommended that patients with an acute EDH in coma (GCS score <9) with anisocoria undergo surgical evacuation as soon as possible. |
| * An EDH less than 30 cm3 and with less than a 15-mm thickness and with less than a 5-mm MLS in patients with a GCS score greater than 8 without focal deficit can be managed nonoperatively with serial CT scanning and close neurological observation in a neurosurgical center. | ||
| Acute subdural hematoma | * An acute SDH with a thickness greater than 10 mm or an MLS greater than 5 mm on CT scan should be surgically evacuated, regardless of the patient’s GCS score. | In patients with acute SDH and indications for surgery, surgical evacuation should be performed as soon as possible. |
| * All patients with acute SDH in coma (GCS score less than 9) should undergo ICP monitoring. | ||
| * A comatose patient (GCS score less than 9) with an SDH less than 10 mm thick and an MLS than 5 mm should undergo surgical evacuation of the lesion if the GCS score decreased between the time of injury and hospital admission by 2 or more points on the GCS and/or the patient presents with asymmetric or fixed and dilated pupils and/or the ICP exceeds 20 mm Hg. | ||
| Traumatic parenchymal lesions | * Patients with parenchymal mass lesions and signs of progressive neurological deterioration referable to the lesion, medically refractory intracranial hypertension, or signs of mass effect on CT scan should be treated operatively. | Bifrontal decompressive craniectomy within 48 hours of injury is a treatment option for patients with diffuse, medically refractory posttraumatic cerebral edema and resultant intracranial hypertension. |
| * Patients with GCS scores of 6 to 8 with frontal or temporal contusions greater than 20 cm3 in volume with MLS of at least 5 mm and/or cisternal compression on CT scan, and patients with any lesion greater than 50 cm3 in volume should be treated operatively. | ||
| * Patients with parenchymal mass lesions who do not show evidence for neurological compromise, have controlled ICP, and no significant signs of mass effect on CT scan may be managed nonoperatively with intensive monitoring and serial imaging. | ||
| Posterior fossa mass lesions | * Patients with mass effect on CT scan or with neurological dysfunction or deterioration referable to the lesion should undergo operative intervention. Mass effect on CT scan is defined as distortion, dislocation, or obliteration of the fourth ventricle, compression or loss of visualization of the basal cisterns, or the presence of obstructive hydrocephalus. | In patients with indications for surgical intervention, evacuation should be performed as soon as possible because these patients can deteriorate rapidly, thus worsening their prognosis. |
| * Patients with lesions and no significant mass effect on CT scan and without signs of neurological dysfunction may be managed by close observation and serial imaging. | ||
| Depressed cranial fractures | * Patients with open (compound) cranial fractures depressed greater than the thickness of the cranium should undergo operative intervention to prevent infection. | Early operation is recommended to reduce the incidence of infection. |
| * Patients with open (compound) depressed cranial fractures may be treated nonoperatively if there is no clinical or radiographic evidence of dural penetration, significant intracranial hematoma, depression greater than 1 cm, frontal sinus involvement, gross cosmetic deformity, wound infection, pneumocephalus, or gross wound contamination. | ||
| * Nonoperative management of closed (simple) depressed cranial fractures is a treatment option. |
EDH, epidural hematoma; GCS, Glasgow Coma Scale, MLS, midline shift; CT, computed tomographic; SDH, subdural hemotoma; ICP, intracranial pressure
References
14. Paczynski RP. Osmotherapy. Basic concepts and controversies. Crit Care Clin 1997;13:105–129
18. Schrot RJ, Muizelaar JP. Mannitol in acute traumatic brain injury. Lancet 2002;359:1633–1634
24. Feig PU, McVurdy DK. The hypertonic state. N Engl J Med 1977;297(26):1444–1454
29. Schwarz S, Schwab S, Bertram M, et al. Effects of hypertonic saline hydroxyethyl starch solution and mannitol in patients with increased intracranial pressure after stroke. Stroke 1998;29(8):1550–1555
63. Taneda M, Kataoka K, Akai F, et al. Traumatic subarachnoid hemorrhage as a predictable indicator of delayed ischemic symptoms. J Neurosurg 1996;84(5):762–768
86. Raichle ME, Plum F. Hyperventilation and cerebral blood flow. Stroke 1972;3:566–575
92. Reivich M. Arterial PCO2 and cerebral hemodynamics. Am J Physiol 1964;206:25–35
103. Cormio M, Gopinath SP, Valadka AB, Robertson CS. Cerebral hemodynamic effects of pentobarbital coma in head-injured patients. J Neurotrauma 1999;16:927–936
138. Davis LE, Baldwin NG. Brain abscess. Curr Treat Options Neurol 1999;1:157–166
139. Calfey DP, Wispelwey B. Brain abscess. Semin Neurol 2000;20:353–360
140. Mathisen GE, Johnson JP. Brain abscess. Clin Infect Dis 1997;25:763–781
147. Grady MS. Decompressive craniectomy. J Neurosurg 2006;104(4):467–468; discussion 468
< div class='tao-gold-member'>









