Fig. 6.1
MARK/Par-1 domains and major modes of regulation. MARK/Par-1 kinases consist of an N-terminal domain (N) a catalytic domain followed by a common docking domain-like CD-like motif, an auto-regulatory UBA domain, a spacer, and a tail domain (or kinase associated domain, KA1). MARK/Par-1 is subject to multiple means of regulations that occur at several sites along the protein. One requirement for MARK/Par-1 activity is the phosphorylation of a threonine at the activation loop (human MARK/Par-1 T208), which keeps the active site accessible to the substrate. The kinases MARKK (TAO1) and LKB1 activate MARK/Par-1 by phosphorylating it on T208 (green arrows). GSK3b inactivates the kinase by targeting a different residue in the same domain, Serine 212 (red blocked lines). CaMKI also targets the catalytic domain of MARK/Par-1. aPKC phosphorylation sites lies within the space domain and its phosphorylation allows the inhibitory binding of 14-3-3 to MARK/Par-1. This binding represents a second mode of regulation of MARK/Par-1, which involves protein-protein interaction (Black blocked lines). 14-3-3 binds to the spacer domain and relocalizes the kinase while PAK5 binds MARK/Par-1 at the catalytic domain itself. Intra-molecular inhibitory interaction (red arched lines) of the tail may block the catalytic domain. Alternatively, the tail may mediate binding of inhibitory partners. The regulatory role of the UBA domain is not fully recognized. Its homology to the UBA class of ubiquitin-associated proteins suggests that it may be able to interact with different forms of polyubiquitin. The common docking domain-like CD-like may exert its function by binding to yet uncharacterized cofactors (The scheme is adopted from Marx et al. 2010)
MARK/Par-1 acts as a key organizer of microtubule arrays that govern polarized transport in both vertebrate (Cohen et al. 2004a, b; Suzuki et al. 2004) and invertebrate (Doerflinger et al. 2003) epithelia. It was identified as an important microtubule organizer in the leading edge of migrating cells downstream of Rac1 activity (Nishimura et al. 2012). The microtubule-associated proteins (MAPs) tau and MAP2 are well-known substrates of MARK/Par-1 (Drewes et al. 1997). MARK/Par-1 phosphorylates these MAPs at their corresponding KXGS motifs, which reside within their microtubule-binding domain, resulting in a dramatic reduction in their affinity to microtubules. The detachment of MAPs from the microtubules can lead to highly dynamic and unstable microtubules (Drewes et al. 1997, 1998). Tau, an axonal MAP, regulates not only microtubules stability but also axonal transport and has a pivotal role in neuronal function and survival (Matenia et al. 2012; Timm et al. 2011; Sydow et al. 2011; Li et al. 2011), as well as regulation of dendritic functions (Ittner et al. 2010). Tau is well known for its involvement in a group of neurodegenerative diseases collectively known as tauopathies [reviewed in (Hernandez and Avila 2007; Avila et al. 2004; Mandelkow and Mandelkow 1998; Rademakers et al. 2004)]. The most common tauopathy is Alzheimer’s disease where hyperphosphorylated tau accumulates within paired helical filaments. During disease progression, hyperphosphorylated tau is missorted to the dendrites and cytoplasm and aggregates into neurofibrillary tangles (NFTs), a process that lead to neuron dysfunction and neurodegeneration (Thies and Mandelkow 2007). NFTs are a pathological hallmark of Alzheimer’s disease (AD) as well as a collection of other neurodegenerative disorders that are referred to as tauopathies [reviewed in (Morris et al. 2011; Morfini et al. 2009; Hernandez and Avila 2007; Avila et al. 2004)]. Based on the recent findings of a dendritic function of tau and studies in mouse models, the role of tau in AD pathogenesis was revisited and it was placed in the amyloid-ß toxicity cascade (Morris et al. 2011). In addition, mutations within the human MAPT (tau) locus result in the neurodegenerative disease, frontotemporal dementia with Parkinsonism (Hutton et al. 1998; D’Souza et al. 1999). However, tau pathologies are not confined to neurodegenerative diseases. Microdeletions of a region encompassing the MAPT gene result in moderate intellectual disability with associated dysmorphic features (Shaw-Smith et al. 2006; Sharp et al. 2006; Koolen et al. 2006, 2008; Varela et al. 2006). The frequency of this microdeletion syndrome was estimated to be 1:13,000 to 1:20,000, thus suggesting it to be a common underlying cause for intellectual disability. When the same area on chromosome 17 is duplicated, patients exhibit behavioral problems and poor social interactions that are consistent with autism spectrum disorders (ASD) (Grisart et al. 2009; Kirchhoff et al. 2007). Interestingly, MARK1, one of the members of the MARK/Par-1 gene family has been suggested as a susceptibility gene for autism (Maussion et al. 2008).
3 MARK/Par-1 Regulation
MARK/Par-1 activity is regulated by several mechanisms [see Fig. 6.1 and review (Marx et al. 2010)]. The kinase can be activated by phosphorylation on a conserved threonine in the activation loop, which can be executed by MARKK/TAO-1 (Timm et al. 2003) or LKB1 in association with STRAD and MO25 (Lizcano et al. 2004). Phosphorylation of an adjacent serine residue by GSK3β resulted in inhibition of MARK2/Par-1 activity in one study (Timm et al. 2008a) or activation in another study (Kosuga et al. 2005). Phosphorylation at other sites may affect MARK2/Par-1 activity or its interactions with additional proteins. For example, phosphorylation by aPKC induced binding of MARK/Par-1 to the scaffold protein 14-3-3/PAR-5, which resulted in relocation of the kinase from the cell membrane to the cytosol (Suzuki et al. 2004; Watkins et al. 2008; Hurov et al. 2004). In addition, several protein interactions regulate MARK/Par-1 activity independent of protein phosphorylation. For example, MARK/Par-1 interaction with PAK5 inhibited the activity of the kinase (Matenia et al. 2005). Furthermore, the C-terminal tail of MARK/PAR1 kinases (which includes the KA1 domain) is involved in binding to acidic phospholipids, which may be important for the interaction with subcellular fractions of the plasma and may also be involved in regulation of enzymatic activity (Moravcevic et al. 2010; Goransson et al. 2006; Hurov et al. 2004). Tight control of MARK/Par-1 may be achieved in part by its regulated degradation; it has been shown that active MARK/Par-1 phosphorylated by LKB1 is targeted for ubiquitination and degradation (Lee et al. 2012).
4 MARK/Par-1 in Primary Neurons
MARK/Par-1 is required for establishment of neuronal polarity in culture (Biernat et al. 2002) (Fig. 6.2). Within the axon, MARK/Par-1 mediated phosphorylation may interfere with the maintenance of the barrier-mediated sorting in the initial axonal segment (Li et al. 2011). Knockdown of MARK/Par-1 induced formation of multiple axons in hippocampal neurons, whereas strong MARK/Par-1 expression inhibited axon formation. This inhibition was counteracted by the Par-3/Par-6/aPKC complex, which inhibits MARK/Par-1 (Chen et al. 2006). Recent research has demonstrated that Par-3 is also involved in regulation of microtubule stability. Furthermore, disruption of the microtubule regulatory activity of Par-3 impaired its function in axon specification (Chen et al. 2013). At least part of the functions of MARK/Par-1 in the axon may be attributed to the activities of one of its main substrates, tau [reviewed in (Matenia and Mandelkow 2009; Timm et al. 2006)]. Tau is mainly an axonal protein (Kempf et al. 1996; Mandell and Banker 1996) however, it is highly dynamic and its axonal sorting is regulated in part by active phosphorylation, for example by MARK/Par-1 (Li et al. 2011; Konzack et al. 2007). It should be noted that tau may be phosphorylated by additional kinases such as GSK-3β and CDK5 [reviewed in (Billingsley and Kincaid 1997; Drewes 2004)]. In vitro work also implicates GSK3β as one of the critical regulators of neuronal polarity [reviewed in (Polleux and Snider 2010)]. Experiments using several types of GSK-3 inhibitors indicate that GSK-3α/β act as negative regulators of axon formation because they lead to formation of multiple axons (Jiang et al. 2005; Yoshimura et al. 2005). The activity of MARK/Par-1 is not limited to axons, although high activity, which was visualized by a cellular biosensor, was noticed in the axon and growth cone of developing neurons (Timm et al. 2011). The active kinase was shown to inhibit the formation of dendrites via phosphorylation of MAP2 in hippocampal neurons (Terabayashi et al. 2007). A role for MARK/Par-1 has also been demonstrated in dendritic spines (Wu et al. 2012). This activity was mediated through MARK/Par-1 induced phosphorylation of the synaptic scaffolding protein PSD-95 (Fig. 6.2).
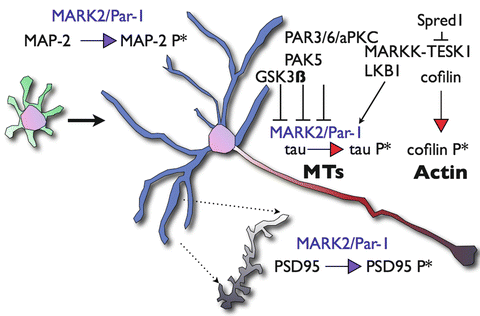
Fig. 6.2
Schematic representation of key substrates of MARK/Par-1 during neuronal polarization. Neuronal polarization of hippocampal neurons in vitro involves the transition from a non-polarized cell extending multiple undifferentiated neurites (green processes) to well-characterized cellular compartments; a single axon (red) and multiple dendrites (blue). Tau is the key microtubule-associated protein in axons and MAP2 is the key microtubule-associated protein in the dendrites. MARK/Par-1 phosphorylation alters their affinity the microtubule and affects microtubules dynamics. The Par3/Par6/aPKC complex counteracts excessive activity of MARK/Par-1 and relives possible inhibition of axonogenesis. In parallel, modulation of the actin cytoskeleton occurs upstream of MARK/Par-1. MARKK an activator of the kinase binds Sprouty-related protein with EVH-1 domain1 (Spred1) and the later interacts and inhibits specific protein kinase (TESK1) thus limiting the phosphorylation of Cofilin, which results in the stabilization F-actin. Phosphorylation of MAP2 by MARK/Par-1 shortens the length and reduces the branching of dendrites. At the mature dendrite, MARK/Par-1 regulates spine morphogenesis through phosphorylating of Postsynaptic Density Protein 95 (PSD-95)
MARK/Par-1 is likely to be mediating an active crosstalk between the microtubules and actin microfilaments [reviewed in (Timm et al. 2006; Matenia and Mandelkow 2009)]. Neuronal polarization requires a continuous modulation of both actin microfilaments and microtubules [reviewed in (Witte and Bradke 2008; Arimura and Kaibuchi 2007)]. The regulatory interactions between MARK/Par-1 and several actin modulators are capable of mediating this crosstalk [reviewed in (Matenia and Mandelkow 2009)]. PAK5, a member of the Rac/Cdc42-associated Ser/Thr kinases inhibits MARK/Par-1 activity resulting in stable microtubules and in parallel induces actin dynamics (Matenia et al. 2005) (Fig. 6.2). A similar type of regulatory interactions occurs more upstream, one of the MARK/Par-1 activating kinases, MARKK has been found to interact with TESK1 and Spred-1 (Johne et al. 2008). TESK-1 belongs to the LIM kinase family and stabilizes actin organization by phosphorylating Cofilin. TESK1 can also bind and inactivate MARKK. Spred-1’s interaction with TESK1, inhibits TESK1 kinase activity. Thus, the tripartite interaction of MARKK-TESK1 and Spred-1 can influence both the microtubules and the actin elements of the cytoskeleton.
In vivo, kinases belonging to the MARK family (SAD-A and SAD-B also known as BRSK1 and BRSK2) have been shown to regulate neuronal polarity (Kishi et al. 2005). Cortical neurons doubly knockout for SAD-A and SAD-B kinases were unable to specify a single neurite to become an axon in vivo. Additional studies demonstrated that the kinase LKB1 is the main upstream activator of SAD-A/B kinases in cortical neurons (Barnes et al. 2007).
5 MARK/Par-1 and Neuronal Migration
MARK2 is a member of a small family of proteins (Tassan and Le Goff 2004), thus, although it could be expected that knockout of the Mark2 gene will result in a neuronal migration phenotype based on its cellular activities, developmental gene redundancy may explain why such a phenotype has not been described in the Mark2 deficient mice. In utero electroporation has been proven to be an efficient way to circumvent gene redundancy, as previously demonstrated in case of the Dcx family of proteins (Bai et al. 2003; Koizumi et al. 2006) [reviewed in (Reiner et al. 2012)]. However, Mark2 knockout mice do exhibit impairments in spatial learning and memory (Segu et al. 2008). Based on the tight correlation between abnormal neuronal migration and mental retardation, it may be hypothesized that these mice exhibit a subtle previously unappreciated phenotype in the developing brain.
During normal brain development, neuroblasts proliferate in the ventricular (VZ) and the subventricular (SVZ) zones (Fig. 6.3a). Following their postmitotic division, neurons adopt a multipolar morphology (Tabata and Nakajima 2003) from which they will transit to a bipolar morphology and migrate along radial glia (Fig. 6.3a, b). Once they reach the cortical plate (CP), they detach from the radial glia and form appropriate connections. Overexpression of MARK/Par-1 in the developing brain using in utero electroporation strongly inhibited neuronal migration; cells lost their polarity and adopted round rather than multipolar or bipolar morphologies (Fig. 6.3c, d). Reduction in the levels of polarity kinase MARK/Par-1 by shRNA resulted in a pronounced inhibition of radial neuronal migration (Sapir et al. 2008a) (Fig. 6.3e, f). Most of the shRNA treated neurons were stalled at the boundary between the intermediate zone (IZ) and CP (Fig. 6.3e, f). The inhibited neurons mainly exhibited a multipolar morphology. Some of the neurons that managed to migrate towards the CP, exhibited abnormal morphology with a curved or bifurcated leading edge pointing to the VZ (Sapir et al. 2008a) (Fig. 6.3f). The transient multipolar stage is sensitive to the levels of quite a few proteins; including DCX, LIS1, Filamin A and others (LoTurco and Bai 2006). Schematic presentation of brain sections treated with Dcx shRNA is shown in Fig. 6.3g, h. The proteins mentioned above regulate cell polarity and motility in neocortical SVZ and IZ during radial migration. Interestingly, adding MARK/Par-1 kinase-dead on top of MARK/Par-1 shRNA allowed neurons to change their morphology from multipolar to bipolar. Nevertheless, this transition was not sufficient to allow successful migration to the CP. This result strongly suggests that there may be different modes of regulation for each phase. This was the first example where a transition to the bipolar morphology occurred without subsequent migration. These results strongly suggest for an unknown kinase-independent activity involved in morphology change. Reduction of kinase activity on its own also retarded neuronal migration and cells did not reach their expected position in the cortical plate. Reduction of kinase activity was achieved by expression of MARK/Par-1 kinase-dead which acts as a dominant negative and inhibits endogenous kinase activity, or via expression of PAK5, which binds to the catalytic domain of MARK2 and inhibits its activity (Matenia et al. 2005).
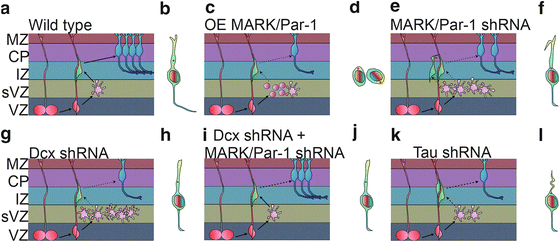
Fig. 6.3
MARK/Par-1 and neuronal migration. (a) Simplified scheme of the radial migratory path of excitatory neurons in the developing cerebral cortex. Cells born at the ventricular zone (VZ) from a radial progenitor (red) one of several types that occupy the subventricular zone (sVZ). The postmitotic cells lose their polarity and become multipolar (pink) before re-establishing a bipolar morphology and moving to the forming cortical plate (CP), sending connections to the marginal zone (MZ) and towards other brain areas. A migrating cell typically orients a leading edge towards the pial surface and the centrosome is located in front of the nucleus and serves as a microtubule organization center. (b–j) MARK/Par-1 affects neuronal migration via its dual role in regulating cellular polarity as well as microtubule dynamics. Over expression of MARK/Par-1 causes destabilization of the microtubule, complete loss of polarity cell rounding and inhibition in radial migration (d). Acute reduction in MARK2/Par-1 levels in the developing brain is also detrimental to normal migration. Neurons treated with MARK/Par-1 shRNA fail to migrate beyond the IZ and fail to repolarize. The leading edge of the migrating neurons is frequently hooked and distorted and the centrosomal motility is decreased (f). Knock down of DCX, a MARK/Par-1 substrate, cause cells to stall in a highly branched multipolar morphology (g) Bipolar cells occasionally display centrosome split and irregular centrosomal motility (h). (i) Dcx shRNA with additional reduction in MARK/Par-1 levels partially rescues this phenotype and restores centrosomal motility (j). (k) Introduction of tau shRNA in the developing brain inhibits neuronal migration. Less neurons reach the cortical plate. The leading edge of the migrating neurons is curved and thin (l)
Taking into consideration the important role that MARK/Par-1 plays in migration of radially migrating neurons, we also examined its role in migration of postnatal neurons to the olfactory bulbs (Mejia-Gervacio et al. 2011). Rostral migratory stream (RMS) neuroblasts expressed MARK/Par-1 in the postnatal and adult brain. Knockdown of MARK/Par-1 revealed that it is required for correct positioning of the leading processes of the neuroblasts heading toward the olfactory bulb. Decreasing the expression levels of MARK/Par-1 in neuroblasts impaired the ability of cells to maintain a sustained forward direction of displacement in the RMS. As a consequence, the integration of newborn neurons into the olfactory bulb circuit was compromised.
It is likely that the observed phenotypes can be attributed at least in part to modulation of microtubule dynamics. MARK/Par-1 phosphorylates multiple substrates, some of them are MAPs, which change their affinity to microtubules following MARK/Par-1 phosphorylation. The substrates include tau, MAP2, MAP4 and DCX (Drewes et al. 1997; Biernat et al. 1993; Schaar et al. 2004). Overexpression of MARK/Par-1 in cells leads to hyperphosphorylation of MAPs on KXGS motifs and to disruption of the microtubule array (Drewes et al. 1997). As mentioned above, in vivo overexpression of MARK/Par-1 resulted in loss of neuronal polarity (Sapir et al. 2008a). Reduction in MARK/Par-1 resulted in more stable microtubules detected in primary neurons, and as a consequence in vivo neurons were stalled in the multipolar stage (Sapir et al. 2008a). Reduction in DCX, one of the substrates of MARK/Par-1, resulted in an opposite effect with more dynamic microtubules, yet in vivo neurons are stalled in the multipolar stage (Sapir et al. 2008b) as previously reported (Bai et al. 2003; Ramos et al. 2006) (Fig. 6.3g). One clear conclusion from the above-described experiments may be that proper neuronal migration requires very accurate control of microtubule dynamics. Tipping the balance in either direction inhibits neuronal migration. Based on this conclusion it was possible to postulate that simultaneous reduction of both DCX and MARK/Par-1 will allow for proper neuronal migration. Indeed, in utero electroporation of both DCX and MARK/Par-1 shRNA resulted in a partial rescue of neuronal migration. These results have practical implications following the clear demonstration that a reduction in the levels of one gene may ameliorate the phenotype observed in case of mutation of another gene in the same pathway.
Previous studies indicated that centrosomal motility and the coupling between the centrosome and nucleus through microtubules is important for neuronal migration [reviewed by (Solecki et al. 2006; Tsai and Gleeson 2005; Vallee and Tsai 2006)]. Centrosomal motility requires the activity of molecular motors and cytoskeletal integrity. Furthermore, this process is subject to a delicate balance of opposing activities thus suggesting that reversible post-translational modifications are likely to be involved in the regulation of dynamics of centrosomal motility and neuronal migration. When MARK/Par-1 levels were reduced, centrosomes moved very slowly (Sapir et al. 2008a) (Fig. 6.3f). The dynamics of observed centrosomal behaviour when DCX was reduced differed markedly (Sapir et al. 2008b). The centrosome separated into two centrioles, which moved bi-directionally and fast (Fig. 6.3h). It has been proposed that the mammalian interphase centrosome consists of two independent units held together primarily as a result of the dynamic properties of the microtubule cytoskeleton (Jean et al. 1999). Furthermore, the balanced activities of kinases and phosphatases play an instructive role in centrosomal splitting (Meraldi and Nigg 2001). Therefore, it is possible to assume that reversible phosphorylation may also be involved in splitting of the centrosome in migrating neurons. DCX is a phosphoprotein and is dephosphorylated by phosphatases. The role of DCX kinases in neuronal migration has been well established, but phosphatases are likely to be as important. Of particular interest is the role of Protein Phosphatase 1 (PP1) in centrosomal splitting (Mi et al. 2007), since this phosphatase is capable of dephosphorylating DCX in a site-specific manner (Shmueli et al. 2006; Bielas et al. 2007). Nevertheless, the role of actin and the associated molecular motor myosin in the maintenance of centosomal integrity cannot be neglected. A basic role of the actin cytoskeleton in centrosomal splitting has been established (Euteneuer and Schliwa 1985; Thompson et al. 2004; Uzbekov et al. 2002). In migrating neurons the role of actin remodelling and the activity of myosin has proven to be essential for proper nuclear and cellular motility (Bellion et al. 2005; Schaar and McConnell 2005; Tsai et al. 2007; Ma et al. 2004). Both DCX and MARK/Par-1 are capable of mediating a crosstalk between the microtubule and actin cytoskeleton (Tsukada et al. 2003, 2005; Matenia et al. 2005; Johne et al. 2008). In summary, seamless motility of the polarized centrosome requires a tight balance of factors involved in regulation of the molecular motors and the cytoskeleton.
As mentioned above, tau is one of the key substrates of MARK/Par-1. Reduction of tau clearly inhibits neuronal migration in the developing cortex, thus revealing a clear developmental role for tau (Sapir et al. 2012) (Fig. 6.3k, l). Reduced tau levels affected the morphology of the leading edge in spite of normal morphology of radial glia (Fig. 6.3l). The leading edge of cells treated with shRNA to tau was crooked and thinner than in control cells. Furthermore, tau has a role in regulation of mitochondria in the migrating cells; both intracellular mitochondrial transport and morphology were severely affected following tau knockdown. Neurons that did reach the CP, were not entirely normal. They exhibited smaller cell somas, far less developed dendrites and a striking reduction in connectivity.
Collectively, these results demonstrate that balanced levels of MARK/Par-1, relative levels of at least two of its important substrates DCX and tau, as well as their phosphorylation status, are of importance in regulation of neuronal polarity, migration and later on connectivity in the developing brain.
6 Concluding Remarks
MARK/Par-1 belongs to a functionally conserved family of protein kinases. It plays key roles in several cellular processes including neuronal polarity and migration and it has major roles in more than one aspect of the neurodegenerative cascades in pathologies such as Alzheimer’s disease. The protein is composed of functional domains, which serve as sites for many levels of regulatory modifications and interactions. Indeed the kinase activity is tightly controlled by means of post-translational modifications (phosphorylation), physical interactions, as well as intra- and inter-molecular regulations of the active kinase. MARK/Par-1 exerts its function by phosphorylating several effectors. The phosphorylation of MAPs such as DCX in the developing neuron, tau in the axon, MAP2 in the dendrite, and PSD-95 in the synapse, affect neuronal polarization and neuronal functioning. Its dual effects on polarization and on the dynamic properties of the microtubules make MARK/Par-1 regulated activity pivotal for proper advancement of radial migration in the developing cortex.
Acknowledgements
O.R. is an Incumbent of the Berstein-Mason professorial chair of Neurochemistry. Our research has been supported in part by the Israel Science Foundation (grant no. 47/10), Minerva foundation with funding from the Federal German Ministry for Education and Research, a grant from the Chief Scientist Office at the Israeli Ministry of Health, under the frame of ERA-Net NEURON (DISCover, IMOS 3-00000-6785), Fritz-Thyseen Foundation (grant Az. 10.11.2.161), the Benoziyo Center for Neurological diseases, the Helen and Martin Kimmel Stem Cell Research Institute, and the David and Fela Shapell Family Center for Genetic Disorders Research.
References
Altman J (1969) Autoradiographic and histological studies of postnatal neurogenesis. IV. Cell proliferation and migration in the anterior forebrain, with special reference to persisting neurogenesis in the olfactory bulb. J Comp Neurol 137(4):433–457. doi:10.1002/cne.901370404 PubMed
Angevine JB, Sidman RL (1961) Autoradiographic study of cell migration during histogenesis of cerebral cortex in the mouse. Nature 192:766–768PubMed
Arimura N, Kaibuchi K (2007) Neuronal polarity: from extracellular signals to intracellular mechanisms. Nat Rev Neurosci 8(3):194–205. doi:10.1038/nrn2056 PubMed
Related posts:
 The Impact of JNK on Neuronal Migration
The Impact of JNK on Neuronal Migration
 Spinal Motor Neuron Migration and the Significance of Topographic Organization in the Nervous System
Spinal Motor Neuron Migration and the Significance of Topographic Organization in the Nervous System
 The PAR Polarity Complex and Cerebellar Granule Neuron Migration
The PAR Polarity Complex and Cerebellar Granule Neuron Migration
 Molecular Pathways Controlling the Sequential Steps of Cortical Projection Neuron Migration
Molecular Pathways Controlling the Sequential Steps of Cortical Projection Neuron Migration
 Microtubules and Neurodevelopmental Disease: The Movers and the Makers
Microtubules and Neurodevelopmental Disease: The Movers and the Makers
 Novel Functions of Core Cell Cycle Regulators in Neuronal Migration
Novel Functions of Core Cell Cycle Regulators in Neuronal Migration
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


