Chapter 65 Methods of Cranial Vault Reconstruction for Craniosynostosis
Cranial sutures are essential components in the development of the skull. Nonfunctional sutures during the evolution of the cranial vault and the skull base lead to evolving deformities that may end in neurologic sequel. Although the term craniosynostosis was first used by Bertolotti in 1914, referring to the premature closure of a cranial suture, it was Sommerring who described in 1791 the anatomy of the suture and postulated not only its role in normal skull growth but also the effects of early closure.50 During the 19th century Otto, and later Virchow, asserted that premature closure of sutures (craneostenosis) prevented growth perpendicular to the suture and was accompanied by compensatory growth at others. Premature closure may affect a single suture, but several sutures may be involved and severe deformities can develop, including the orbits and anterior fossa in the process. This is even more evident in craniofacial syndromes, where the main difference between single-suture and nonsyndromic craniosynostosis is the alterations not only in neurocranium but also in viscerocranium, resulting in anomalies in the midface skeleton.
The true incidence of craniosynostosis is not known, but it is estimated to occur in 1 of 2500 newborns. Sagittal synostosis is the most frequent (40%-60% of all cases), followed by metopic, coronal, and lambdoid, which is unusual.1,2 More than 150 associated syndromes have been described, but in most cases craniosynostosis is an isolated phenomenon. Several theories have been postulated to explain the appearance of premature synostosis and posterior deformities. Virchow was the first to suggest in 1852 that the premature closure of the suture was the primary event, while the vault deformity was a consequence of this closure.71 In 1959, Moss suggested that the deformity in the cranial base was the primary event. For this author, dura mater is an important regulator in the activity of the sutures and vault development and the main factor for the alteration in the cranial growth and suture dysfunction. The abnormal mechanical strengths driven through dural structures from certain maldeveloped points at the skull base (crista galli, petrous pyramids, or sphenoidal wings) would be responsible for cranial deformities and suture dysfunction.3 Park and Powers referred to mesenchymal abnormalities in bone structures, related to genetic anomalies, that would explain more thoroughly hypoplasia affecting the craniofacial region in syndromic cases.67,68,70
In the end, all of these mechanisms and changes produce a cosmetic deformity, as well as incompetence of the cranial and facial structures—unable to properly contain the organs inside the vault (brain and cerebellum) and the orbits (optic nerves and eyeballs)—and hypoplasia of the midface and oropharyngeal region. All of these factors are related, and depending on the affected region, there is a predominance of one abnormality. However, as explained later, in craniofacial syndromes the situation is more complicated. The presence of raised intracranial pressure (ICP) (multiple suture closure), hydrocephalus and cerebrospinal fluid (CSF) circulation anomalies, venous hypertension (skull base sutures closure affecting jugular foramina drainage, as well as genetic factors including endothelial proliferation in dural venous sinuses), chronic hypoxia and hypercapnia (obstructive airway in relation to midface retrusion and amygdalar hyperplasia), and chronic tonsillar herniation leads to a complex situation in which staged treatment has to be the rule, particularly after careful and proper understanding of the pathophysiology underlying these cases and always in a multidisciplinary team.
Scaphocephaly
Sagittal synostosis is the most common form of craniosynostosis. The premature closure of the sagittal synostosis has an estimated prevalence of 2 in 1000 births. Up to 80% of the cases are sporadic, and there is a male-to-female ratio of 3:1 to 4:1.1 Sporadic cases are mainly related to intrauterine constraint, but some may have a heterogeneous cause. In particular cases, a dominant autosomal inheritance has been described, with 38% penetrance.1 The frequency of twinning in a series was 4.8%, with only one monozygotic twin pair being concordant for sagittal synostosis.4
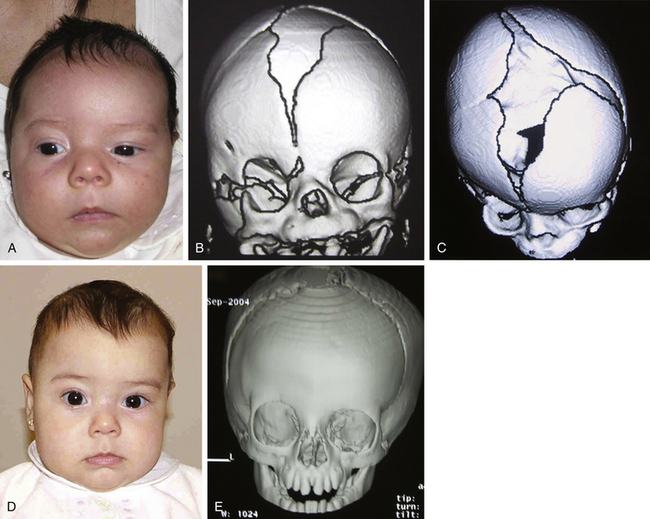
Histology usually shows synostotic ridging on the posterior part of the fused suture, which is consistent with the sagittal suture being thicker on the ectocranial cortex than on the endocranial side, and the bony edge of the suture becomes progressively thicker from anterior to posterior under normal conditions.5 When the sagittal suture is the only involved suture, the head becomes long from front to back (Figs. 65-1 to 65-3). It is narrow from side to side, and frontal compensational bossing may occur when the anterior fontanel is patent. When both fontanels are closed and the posterior part of the suture is predominantly affected, there is a posterior deformity typically referred to as an occipital knob. It is usually accompanied by a thin forehead with severe pterional indentation (leptoscaphocephaly). In the severest forms of scaphocephaly, the head adopts the shape of a saddle, and it is named bathrocephaly. Accurate diagnosis of isolated sagittal synostosis can be made clinically, and operative correction can proceed without a need for radiologic investigations, unless the clinical features are not completely typical.6 This could be the case in which Crouzon syndrome in some patients can resemble, early in evolution, an isolated scaphocephaly.
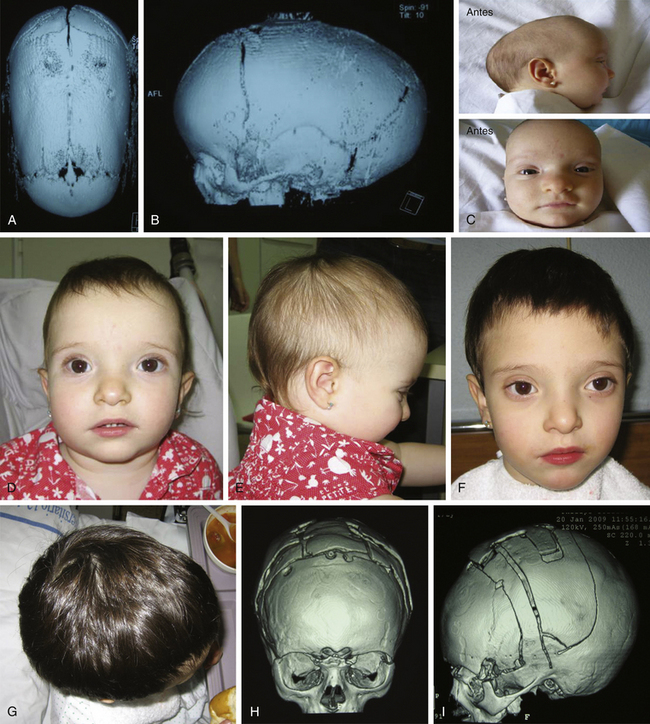
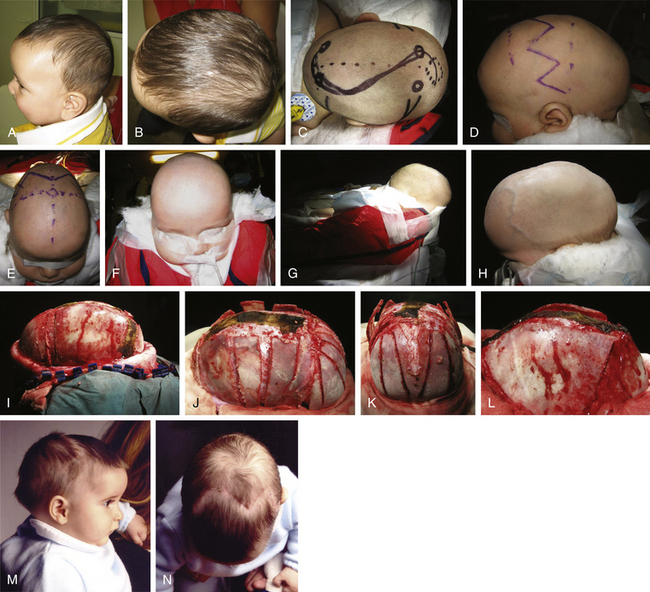
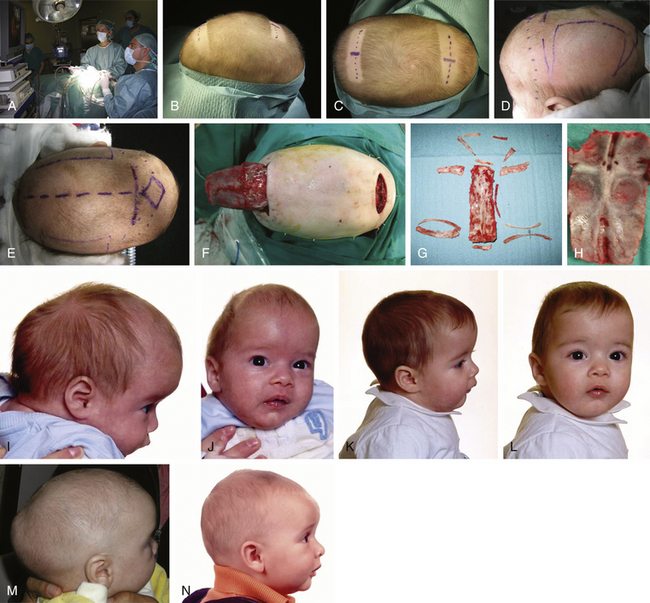
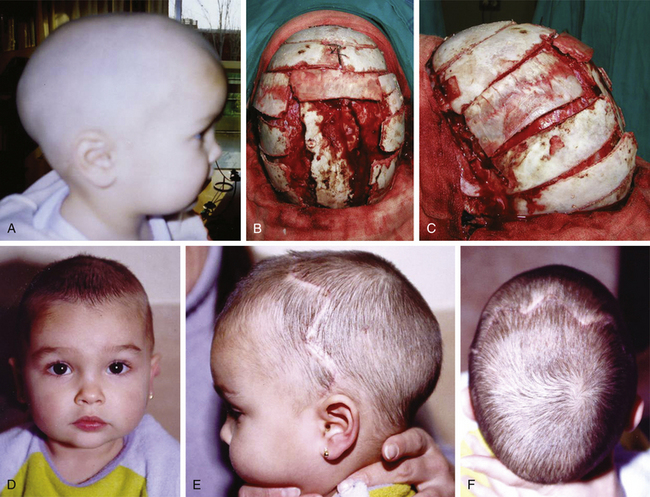
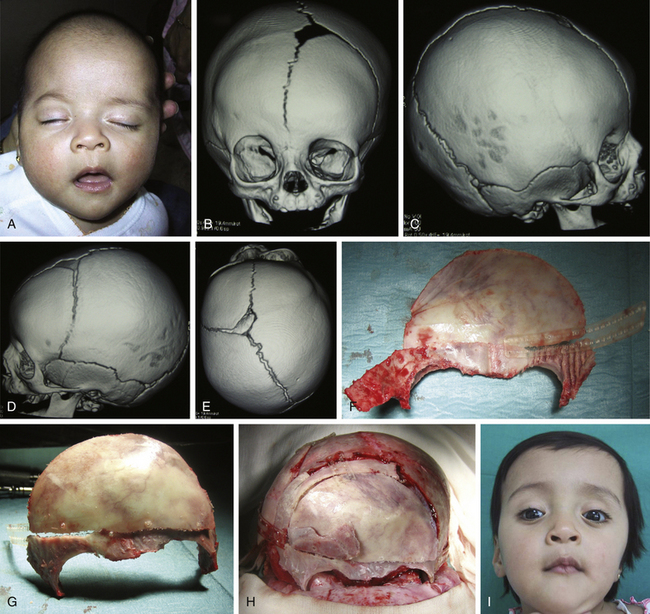
There is a plethora of techniques for the treatment of scaphocephaly7–11 but most of them include the excision of the sagittal suture with different kinds of parietal and occipital osteotomies with or without frontal osteotomies and remodeling. Surgical correction, whatever the technique, when performed early in infancy, results in satisfactory and persisting reshaping of the skull (Figs. 65-1 to 65-3). Delays in diagnosis, as well as treatment, result in a small subset of patients with uncorrected craniosynostosis that require the development of more extensive calvarial reconstruction techniques (Fig. 65-4). Due to calvarial maturity, these more advanced cases would not be amenable to treatment using less aggressive techniques. After 1 year of age the child’s cranial vault has developed abnormally, resulting in a severe skull base deformity, as shown in the figure. Subsequently, the skull is significantly thicker and less pliable to reshaping. At this age, cranial defects due to aggressive surgical remodeling procedures may not reossify, and there is a lack of the driving mechanism that leads to closure of the sutures. For these patients, an approach with a more comprehensive calvarial reconstruction includes multiple interleaving osteotomies crossing the midline, leading to improvements in biparietal narrowing combined with a bifrontal reconstruction.
Trigonocephaly
Trigonocephaly is considered a fairly common type of craniosynostosis. The term was first coined by Welcker in 1862.12 Formerly denoted as unusual,12,13 it is now considered to represent about 10% of all patients with deformities related to craniofacial centers12,14,15 and its frequency is increasing.2 Its incidence is estimated to be 1 in 2500 births,16 and there is a male predominance of 65% to 85%.14
Closure of the metopic suture starts under normal circumstances at the end of the first year and may last until the end of the second year.12 When premature suture closure occurs, usually before birth, a typical craniofacial malformation develops. The premature arrest of growth of the metopic suture may present as a spectrum of manifestations, depending on the timing and extent of the suture closure. Its mildest form is a familial and ethnically inherited facial morphology.17 Milder forms of metopic synostosis consist of only a somewhat prominent metopic ridge that does not need surgical intervention. In severe forms, a characteristic keel-shaped forehead can be observed, with absence of frontal eminences, retruded orbital rims, hypotelorism, and epicanthus giving a peculiar craniofacial appearance to these children. Different degrees of involvement of the anterior chondrocranial structures seem possible, such as presphenoid, mesoethmoid, and ectoethmoid structures.18 Another explanation for the broad phenotype of affected children could be a nonspecific process acting at different evolutive ages. Trigonocephaly may appear either as an isolated anomaly or as part of syndromes involving prosencephalic or rhinencephalic structures (holoprosencephaly), such as Opitz syndrome,19,20 Say-Meyer syndrome, or Frydman syndrome.12 Recently a new fronto-ocular syndrome has been described with trigonocephaly.21
Several genetic defects also have been described associated with trigonocephaly. Abnormalities including deletions and duplications of chromosomes 3p, 9p, 1, lq, and 13q have been published.22–25 Isolated trigonocephaly has an unknown etiology. Autosomal dominant inheritance with very low penetrance has been proposed in 2% to 5% of the cases for familial cases.15,26 The possible involvement of mutated FGFRs in newborns affected by trigonocephaly has recently been investigated by molecular screening in a search for mutations in FGFR2, which has been implicated in complex craniofacial syndromes. However, none of the cases studied carried mutations, except one that evolved later toward a Crouzon syndrome–like profile.25
Mental delay may occur in isolated trigonocephaly and has been observed in as many as 10% of these children.12 Development delays may consist of subtle changes in the time of acquisition of head control or sitting position or include more severe forms of mental retardation, with or without intracranial hypertension.
Di Rocco et al. proposed two subgroups on the basis of clinical and radiologic findings.18 Group I presents with bilateral frontal bone hypoplasia associated with extreme retrusion of the supraorbital margins. In this group, hypotelorism is associated with abnormally deep position of the cribriform plate, giving the ethmoidal region a hollow appearance. In these patients, the nasion–pterional angle is severely restricted and the nasion–clinoidal distance significantly increased. Group II also shows bilateral frontal hypoplasia with hypotelorism, supraorbital retrusion, and a reduced nasopterional angle. However, the nasion–clinoidal distance is almost normal, and pterional evidence is scarcely evident.18 Moreover, patients in group II showed a lesser degree of temporal compensatory expansion. The authors postulate that in some patients a compensatory elongation of the nasion–clinoidal distance and an incomplete synostosis of the frontoethmoidal sutures allows partial lateral expansion of the anterior cranial fossa, which could diminish the need for posterior calvarial expansion. On the other hand, children with more severe involvement of the nasoethmoidal sutures, resulting in diminished lateral expansion of the anterior fossa, need compensatory changes in the temporal and parietal regions. Patients in group II accomplished good correction of associated hypotelorism, whereas patients in group I did not reach normal interorbital values when treated with the same surgical procedure, which did not include specific treatment of hypotelorism. Other authors have addressed the importance of the moment and degree of involvement of pathologic changes in the anterior chondrocranial structures.14,27–31 Milder forms of trigonocephaly would affect only the upper metopic suture, whereas more severe forms include involvement of presphenoid, mesoethmoid, and ectoethmoid structures.
Surgical Technique
Before starting osteotomies, the obliquity of frontal bones, retrusion of the lateral portions of the orbital bandeau, and pterional indentation are assessed. A bifrontal craniotomy is performed after a supraorbital bandeau is created. The marks are extended from the sphenosquamosal suture to its contralateral equivalent prepared for a tongue-in-groove advancement.18 The bifrontal flap includes the anterior part of the bregmatic fontanelle and both coronal sutures. The anterior fossa is exposed extradurally with visualization of the anterior two thirds of the orbital roof.
There is not yet a consensus on hypotelorism correction. Several papers have addressed the importance of associated hypotelorism17,28 and the need for direct surgical approach. Different authors17,27,28,36,37 propose adding a nasofrontal osteotomy and an interpositional bone graft to the supraorbital bar and nasoethmoidal area to correct hypotelorism simultaneously with lateral orbitary wall expansion17 or three quarter orbital wall osteotomies.27 On the other hand, there are others13,34 who do not advocate direct approach to the hypotelorism in the belief that internasalis grafting widens only nasal bones without increasing interorbital width, as far as osteotomies usually remain anterior and superior to nasoethmoidal complex.36 I recommend a standard approach without grafting and thus consider the latter ineffective and unnecessary. Both techniques have achieved statistically significant improvement in hypotelorism, greater than that expected from normal growth curves. However, undercorrection of hypotelorism and persistence of abnormally low interorbitary distances occur frequently when orbital widening is not addressed.18,27,35
Anterior Plagiocephaly
Premature closure of a single coronal suture produces the most complex set of craniofacial deformities due to the peculiarities of the fronto-orbital region and the close relationship between the sutures of the anterior cranial base and the orbits and maxillary complex. The incidence of coronal craniosynostosis has been reported to be variable but can be approximated to be 0.4 per 1000.40 Although most cases are sporadic, a familiar incidence of up to 8% in uncomplicated coronal synostosis has been reported.40
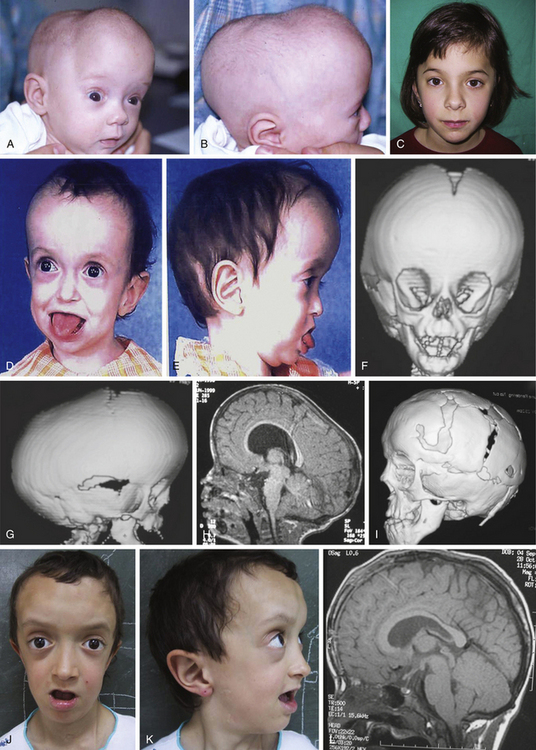
Frontal or anterior synostotic plagiocephaly has been described in association with unilateral coronal synostosis,41–44 but it is known to occur with synostosis of other parts of the coronal ring, such as the frontosphenoidal or frontoethmoidal sutures.41,45–47 In some rare cases, it has been reported to be the result of fusion of the frontosphenoidal48 or the frontozygomatic sutures alone.49 The deformity is manifest as a progressive cranial, orbital, and facial asymmetry of varying severity, with secondary deformities, considered to be compensatory, existing on the contralateral side (Figs. 65-5 and 65-6). Associated cranial anomalies affect the development of all facial bones and have been referred to as “the most complete presentation of craniofacial asymmetry.”50 There is a flat and retruded frontal bone in the affected side with a contralateral bossing. The orbital rim on the synostotic side is also elevated and retruded, while the ipsilateral orbit is elevated and twisted. There is a variable degree of vertical orbital dystopia. The nasal root is deviated toward the closed sutured with an oblique nasal axis. The tip of the nose points to the nonsynostotic side. The zygoma and maxilla are hypoplastic on the affected side, and the ipsilateral temporal bone is malpositioned in an anterior and descended configuration.
Different reconstructive techniques have been described,42,43,46,47,50–53 but most of them can be divided between unilateral and bilateral approaches. There is a long-standing controversy on the timing and type of surgical approach. Some authors have advocated early surgery (younger than 6 months),40,54 whereas others support treating these patients later (between 9 and 10 months).41,45,51 The former group uses the concept of rapid brain growth leading to advancement of the frontal bones and supraorbital rims.
Proponents of late surgery argue that delayed surgery allows for further maturation and growth of the craniofacial skeleton. There is less reliance on brain growth, and blood loss is better tolerated. Also, there is immediate correction of the deformity. Surgical procedures have expanded from simple suturectomies (Fig. 65-5) to frontal–calvarial vault remodeling consisting of bifrontal craniotomes and orbital frontal bandeau advancement (Fig. 65-6). Aggressive surgical approaches have been supported by the theory of “coronal ring” synostosis, which states that there is an extension of the coronal synostosis into the cranial base.55,56 However, in most cases of isolated, nonsyndromic, unilateral coronal synostosis, the sphenofrontal, sphenoethmoidal, ethmoidofrontal, and pterional sutures are patent. Furthermore, review of the literature of these patients who have undergone more extensive procedures indicate that the results are inconsistent and mixed. Often, there is restriction of forward anterior skull base growth and advancement, and late correction (older than 9 months) fails to improve vertical dystopia, which persists into adulthood.40 Most craniofacial centers use the classic fronto-orbital advancement with unilateral or bilateral correction. Undoubtedly, in cases with severe contralateral compensation (frontal and/or temporal bossing), a bilateral correction is preferred. The correction of orbital dystopia warrants careful preoperative evaluation. Orbital hypoplasia is a consequence of the elevated orbital roofs, frontal development of the greater sphenoidal wings, and descent of the ethmoidal position, secondary to early closure of the coronal sutures. Orbital axes are also altered and point downward and outward. In the severest cases, failure to achieve early treatment or undercorrection may lead to strabismus, amblyopia, and anomalies in the binocular vision.
Multiple Suture Craniosynostosis
Brachycephaly
Brachycephaly is most commonly the result of premature synostosis of both coronal sutures. The anterior fossa adopts a characteristic deformation, and there are retruded frontal bones and orbital rims with a vertical, broad, flat forehead and a high bregmatic point. Brachycephaly may appear as an isolated synostosis and has then a favorable prognosis. Often, it is the common final appearance of different syndromic craniosynostoses that share a premature closure of bicoronal sutures and similar phenotype (e.g., some cases of Saehtre-Chotzen or Pfeiffer type I). For this reason, every patient with brachycephaly should be assessed by a clinical geneticist (Figs. 65-7 and 65-8).
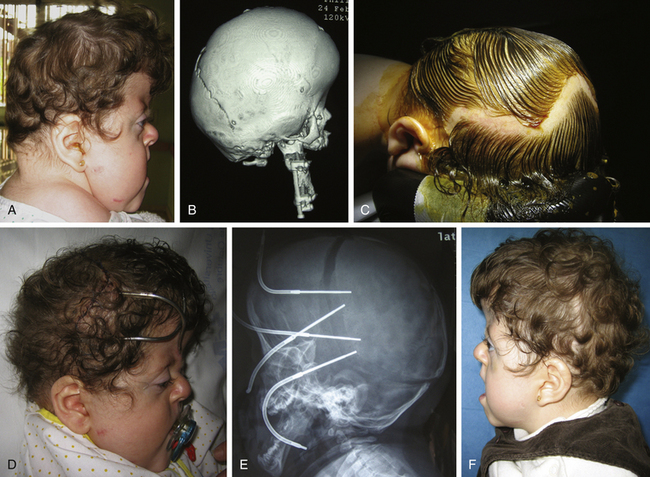
Crouzon Syndrome
Crouzon syndrome is an autosomal dominant disorder characterized by craniosynostosis that causes secondary alterations of the facial bones and facial structure. Common features include hypertelorism, exophthalmos and external strabismus, parrot-beaked nose, short upper lip, hypoplastic maxilla, and a relative mandibular prognathism. First described by Crouzon in 1912,59 it was not until 1959 that Shiller observed an autosomal dominant transmission.60 Crouzon syndrome represents approximately 4.8% of cases of craniosynostosis at birth, and the birth prevalence is estimated to be 16.5 per million births. Crouzon craniofacial dysostosis is linked to a high number of different mutations, but most of them are located on IgIII of FGFR2 (exons 7 and 9) on chromosome 10q. An association between Crouzon syndrome and acanthosis nigricans has been described and is related to an A391E mutation in the FGFR3 gene on chromosome 4p.
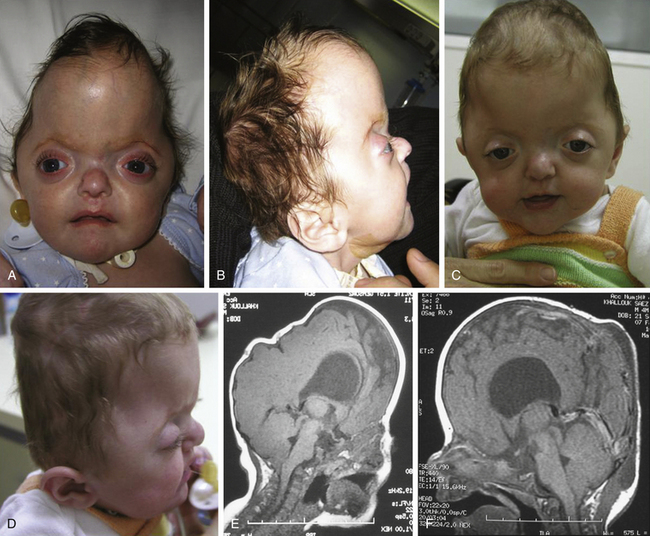
Crouzon syndrome is characterized by a premature synostosis of both coronal sutures, with a resultant brachycephalic shape of the skull. Sagittal, metopic, or lambdoid sutures may also be prematurely affected, isolated, or combined (Figs. 65-9 and 65-10). The cranial base and upper facial sutures are involved with a variable degree of midface hypoplasia and malocclusion. The orbits are hypoplastic, and the orbital floor is hollow, resulting in proptosis and additional orbital dystopia that may produce mild to moderate orbital hypertelorism and divergent strabismus. Maxillary hypoplasia results in pseudoprognatism. Nasal septum deviation, together with maxillary hypoplasia, may produce a chronic obstruction to respiratory flow and may be associated with choanal atresia, velopharyngeal incompetence, and relative macroglossia. All of these malformations may lead to severe respiratory obstructions and apneas, which may cause a chronic raised ICP by an increase in venous pressure after hypercapnia and even contribute as an etiopathogenic mechanism in the development of hydrocephalus.
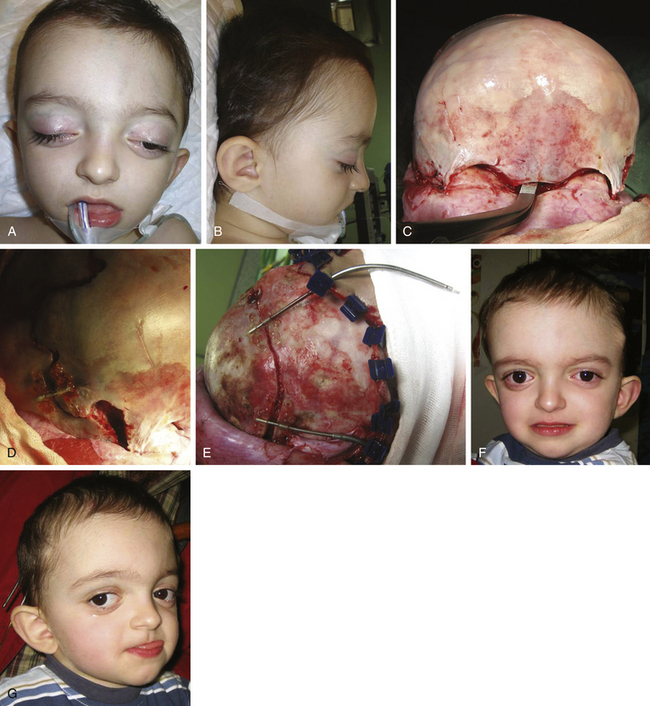
The initial treatment for Crouzon syndrome generally requires cranio-orbital decompression, including bicoronal suture release and osteotomies of the anterior cranial vault and upper orbits with reshaping and advancement. They are usually performed by the age of 8 to 11 months unless signs of increased ICP are found earlier. Sometimes, it is necessary to perform a combined approach, including midface advancement.61 When Chiari malformation (CM) is identified precociously, an occipital–parietal calvarial decompression may be preferable to achieve a bigger expansion of the intracranial volume, with or without suboccipital decompression.62 After proper release of the ICP, fronto-orbital remodeling can be achieved via an anterior approach.
Apert Syndrome
Apert syndrome, also known as acrocephalosyndactyly type I, is a congenital disorder characterized by multiple craniosynostoses, facial hypoplasia, and osseous syndactyly of the hands and feet. Approximately 1 in 65,000 to 165,000 of live births is affected. It is usually classified among a group of craniofacial syndromes with Crouzon, Pfeiffer, and Saehtre-Chotzen syndromes, all of which are allelic disorders with similar clinic manifestations and common genetic background. Wheaton first noted the coincidence of craniosynostosis and syndactyly (1894), but it was Apert who fully characterized the syndrome in 1906.63,64 Apert syndrome consists of a craniosynostosis in the shape of an acrocephaly or a brachycephaly due to the premature closure of several cranial sutures, typically coronal sutures and later those of the anterior cranial base and posterior fossa. The sagittal suture is typically widened and opened. It is always accompanied by osseous syndactyly of hands and feet and by fusion of the distal phalanxes. There is usually severe hypoplasia of the midface with an ogival-fissured hard palate. The orbital rims are retruded and elevated, and the skin possesses a characteristic acneiform appearance, mostly over the nasal bridge, shoulders, and back. Midface anomalies and anterior fossa craniosynostoses produce a decrease in the orbital volume that may lead to proptosis, strabismus (V syndrome), hypermetropia, or astigmatism. Most of these patients present with central nervous system anomalies, including hypoplasia of the corpus callosum and limbic and mesial temporal structure malformations. Approximately 10% of these children develop hydrocephalus, but only 2% of them suffer from a CM, in contrast to Crouzon syndrome, where 75% of the patients develop a hindbrain herniation.61 If left untreated, the incidence of raised ICP has previously been reported as 45%.64,65 Untreated intracranial hypertension may result in insidious optic atrophy, visual loss, and possible developmental delay.
Early cranial decompression with occipital expansion or fronto-orbital advancement is the treatment of choice, although some authors have advocated avoiding routine vault expansion in the first year of life. Instead, careful clinical, ophthalmologic, and respiratory monitoring would allow raised ICP to be treated in the most appropriate manner only when it occurs.65 Midface hypoplasia accounts for the exorbitism, strabismus, and respiratory difficulties seen in this condition. In the absence of any clinical sequelae from this hypoplasia, midface advancement is usually postponed until an age of 4 to 6 years. In cases of functional or clinical compromise, it may be necessary to intervene earlier with different techniques, such as monobloc advancement, maxillotomies, and/or mandible distraction. Facial hypoplasia and ogival palate abnormalities are responsible for phonetic disorders, abnormal dental eruption, and malocclusion.
Apert syndrome patients present with a variable degree of mental retardation. Early treatment of the craniosynostosis has been related to better outcomes. Some authors have shown that up to 50% of patients with Apert syndrome have a normal or near-normal intelligence quotient (IQ), with the rest being moderately to severely retarded. Renier et al.72 found an IQ greater than 70 in 50% of Apert syndrome patients when they were treated by cranial expansion in the first year of life but only in 7.1% of those who were treated later.
Pfeiffer Syndrome
Initially described by Pfeiffer in 1964,67,75 Pfeiffer syndrome is characterized by turribrachycephaly, maxillary hypoplasia, and antimongoloid slant of the orbits. There is hypertelorism and a marked degree of proptosis due to the bicoronal synostosis and subsequent recession of supraorbital rim and short anterior fossa. Extremities are notable for broad thumbs and large toes. There may be a variable degree and number of soft tissue syndactyly (most commonly between the second and the third digits), in comparison to Apert syndrome, where bony syndactyly is the hallmark. There may also be symphalangism (phalangeal fusion), ankylosis of the elbow joints, and cervical vertebral fusions (all of them also possible in Apert syndrome).
There are three clinical subtypes of Pfeiffer syndrome,73 which have clinical and prognostic significance. Type I (classic) is a lesser craniosynostosis that has a typical appearance of brachycephaly but usually is associated with normal or near-normal intelligence. Children with this type of Pfeiffer syndrome can develop normally, but there is often increased ICP if the synostoses are left uncorrected. Type II is associated with cloverleaf deformity, and although compatible with life, prognosis is poor. Type III is characterized by striking proptosis, but kleeblattschädel is absent. Long-term prognosis is poor.
Cloverleaf Skull (Kleeblattschädel)
Kleeblattschädel syndrome is characterized by a trilobar skull caused by frontal and bitemporal bossing. The first description of a trilobar cranial malformation in a newborn was made by Vrolik in 1849,77 but the term kleeblattschädel syndrome was not introduced until 1960 by Holtermuller and Wiederman.77,78 These authors reported a series of children with a trilobar or “cloverleaf” cranium associated with multiple craniosynostoses. In 1965, Commings et al. published the first case in the United States and translated the term kleeblattschädel syndrome into cloverleaf skull.78 This malformation has been described in children with Crouzon, Apert, Carpenter, Beare-Stevenson, or type II Pfeiffer syndrome, the latter being most frequently associated (up to 20% in some series).75,77 The etiology of this syndrome is still unknown and has been attributed to abnormalities of both the calvarium and the skull base, probably due to the presence of a combination of prematurely fused cranial sutures and hydrocephalus.
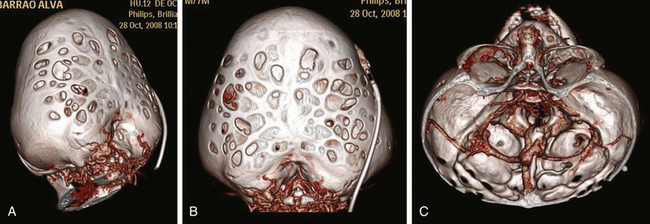
The surgical management of cloverleaf skull remains one of the most formidable challenges for the craniofacial surgeon (Fig. 65-8). The natural history of this deformity without surgery was well described in the 1960s and 1970s. Just a few of these children survived beyond infancy, and those who did developed severe neurologic deficits. The first attempt at surgical correction was reported by Angle in 1967.77 Since then, a number of surgical series have been reported, without a clear conclusion on prognosis and outcome. Review of the literature reveals a lack of consistent surgical strategies.74–76 Postoperative data are sparse, and reoperation is the rule in these patients, with a consequent increased morbidity. In addition to the significant cosmetic deformity, the constant coexistence of intracranial hypertension, hydrocephalus, hindbrain herniation, severe skull base dysplasia, and anomaly in venous drainage (Fig. 65-11) imposes important neurosurgical considerations throughout treatment.
< div class='tao-gold-member'>
Related posts:
 Cortical and Subcortical Brain Mapping
Cortical and Subcortical Brain Mapping
 Radiation Therapy and Radiosurgery in the Management of Craniopharyngiomas
Radiation Therapy and Radiosurgery in the Management of Craniopharyngiomas
 Surgical Management of Intracerebral Hemorrhage
Surgical Management of Intracerebral Hemorrhage
 Neurovascular Decompression in Cranial Nerves V, VII, IX, and X
Neurovascular Decompression in Cranial Nerves V, VII, IX, and X
 Role of Gamma Knife Radiosurgery in the Management of Arteriovenous Malformations
Role of Gamma Knife Radiosurgery in the Management of Arteriovenous Malformations
 Arachnoid, Suprasellar, and Rathke’s Cleft Cysts
Arachnoid, Suprasellar, and Rathke’s Cleft Cysts
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


