and Kelly Del Tredici1
(1)
Zentrum f. Biomed. Forschung AG Klinische Neuroanatomie/Abteilung Neurologie, Universität Ulm, Ulm, Germany
The protein tau is central to the pathological process underlying sporadic AD. It is chiefly a neuronal protein and is produced in all portions of the nervous system (CNS, PNS, ENS) (Trojanowski et al. 1989). In immature nerve cells, tau is distributed diffusely throughout the entire cell, but during cell maturation it converts into a primarily axonal protein. Tau originates in the cell soma’s rough endoplasmatic reticulum, it clears all cellular checkpoints for quality control, and, in this fully functional state, is then transferred to the axonal compartment (Lee et al. 2001; Scholz and Mandelkow 2014). In healthy nerve cells, the protein promotes self-assembly of axonal microtubules and stabilizes them (Fig. 4.1a). Together with motor proteins, microtubules are required for anterograde and retrograde transport of substances within the axon. To a lesser extent, the protein is also present in neuronal somata, dendritic processes, astrocytes, and oligodendrocytes. However, in contrast to the situation in axons, the microtubules in the somatodendritic compartment are mainly stabilized by other microtubule-associated proteins (Mandelkow and Mandelkow 1998, 2012; Binder et al. 2004; von Bergen et al. 2005; Ávila 2006; Goedert et al. 2006; Alonso et al. 2008; Iqbal et al. 2009).
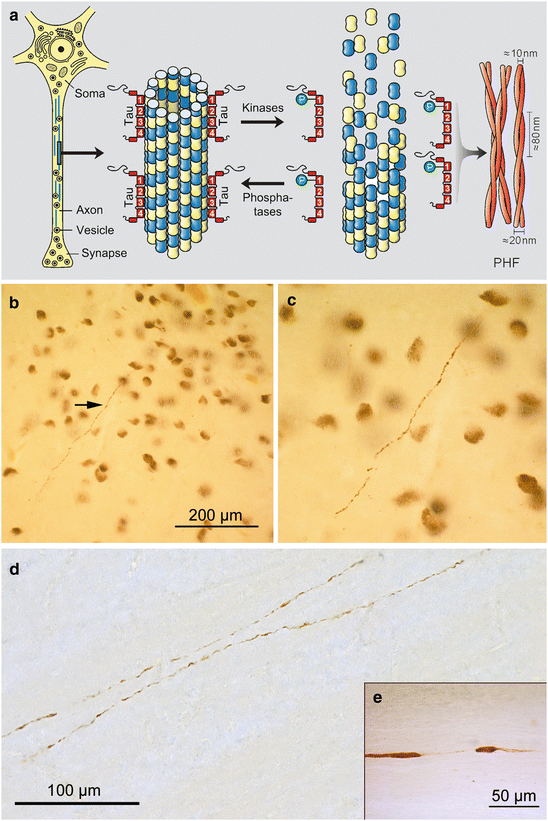
Fig. 4.1
(a) Schematic drawing of axonal microtuble (adapted with permission from EM Mandelkow and E Mandelkow, Trends Cell Biol 1998;8:425–427). (b–d) In stage a, abnormal tau occurs in axons of coeruleus projection neurons. (b) Arrow points to an AT8-ir axonal process in the locus coeruleus of a 14-year-old male shown in (c) at higher magnification. (d) Again, based on their length and location (here, in the superior cerebellar peduncle of a 26-year-old female), these AT8-immunoreactive (AT8-ir) processes are axons rather than dendrites. Note that within both axons non-immunoreactive segments are interspersed by AT8-ir segments (see also insert in e). Abbreviation: PHF paired helical filaments. Scale bar in micrograph (d) applies also to (c)
In the CNS of the human adult, six isoforms of tau are generated from a single gene on chromosome 17q21.31 by alternative mRNA splicing. The isoforms range from 381 to 441 amino acids in length and, in the PNS, they are supplemented by a “big tau” with 695 amino acids. The six isoforms differ by the presence or absence of up to three inserts. In the fetal brain, only the shortest isoform is present and is more extensively phosphorylated than tau in the adult brain. Fetal tau is probably necessary for microtubules during periods of intense plasticity in early brain development. In general, nerve cells of the human adult (including those that become involved during AD) show three-repeat (3R) and four-repeat (4R) isoform levels that are in equilibrium. Other tauopathies, such as progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), and argyophilic grain disease (AGD), exhibit a selective four-repeat accumulation, whereas tau in Pick’s disease (PiD) displays a selective three-repeat isoform (Goedert et al. 1992, 1997; Buée et al. 2000; de Silva et al. 2003; Yoshida 2006; Uchihara et al. 2011, 2012; Niblock and Gallo 2012).
Consistent with its natively unfolded character, a large fraction of the tau protein consists of hydrophilic amino acids, which make it highly flexible and soluble in the cytosol. The protein has numerous binding sites at which it can undergo phosphorylation. Tau’s phosphorylation status is constantly subject to fluctuation: it can transiently be up-regulated—for instance, during mitosis, anesthesia, or hibernation—without resulting in long-term harm to neuronal networks. Hyperphosphorylation of tau also occurs during early developmental phases when it enables axonal growth cones to achieve maximal flexibility for guidance of axons to their target regions and for synapse formation. Thus, an occasional and limited degree of hyperphosphorylation at appropriate sites solely on the part of monomeric tau is not an index of a disease state (Arendt 2004; Run et al. 2009; Seitz et al. 2012; Whittington et al. 2013).
Normal tau binds relatively tightly to axonal microtubules, whereas transiently hyperphosphorylated tau dissociates from microtubles and becomes redistributed freely in the axoplasma. Physiologically, a relatively stable equilibrium emerges between axonal tau microtubule binding and disengagement, thereby indicating a continuous alternation between a less highly phosphorylated state (i.e., bound to axonal microtubules) and a slightly more highly phosphorylated state (i.e., soluble in the axoplasm). This equilibrium of monomeric tau molecules is directed by local axonal kinases and phosphatases (Ballatore et al. 2007).
Monomeric hyperphosphorylated tau is highly soluble and, as such, a large proportion of the protein probably is lost during tissue fixation. It may be for this reason that currently available tau-specific antibodies preferentially mark forms of the protein that already are to some extent aggregated and slightly more viscous than monomeric tau and in this form perhaps more amenable to fixation. All of the various isoforms of tau contain about 30 or more phosphorylation sites (Goedert et al. 1992; Buée et al. 2000). Aggregated and hyperphosphorylated tau protein can be visualized with antibodies, such as the antibody AT8, which recognizes phosphate-dependent epitopes (Mercken et al. 1992). AT8-immunoreactions are highly robust, functioning reliably even in tissue obtained under suboptimal conditions, including longer post-mortal intervals or long tissue storage in aqueous solutions of formaldehyde (Pikkarainen et al. 2010). Thus, AT8 immunohistochemistry is recommended for visualizing abnormal tau during routine neuropathological assessment (Alafuzoff et al. 2006; Montine et al. 2012).
When it is tightly bound to microtubules, monomeric tau is protected against aggregation and, for this reason, only hyperphosphorylated tau species can aggregate. Given an initial conformational change (cross-sheet) followed by some degree of aggregation, the protein tau undergoes conversion from a normal to a pathologically altered cellular component (von Bergen et al. 2005). Here, it should again be emphasized that formation of aggregated tau is an exceptional event that only occurs in a minority of nerve cell types. It is still unclear, however, what the causes for this selectivity are, what conditions need to prevail, and which co-factors have to be present before tau aggregation begins in prediposed types of nerve cells.
One possible precondition for aggregation is a transitory peaking of high concentrations of monomeric hyperphosphorylated tau in the axoplasm. These conditions and other presently unknown factors could abruptly produce initial tau aggregates. The resultant increase in the size of the tau molecules coupled with a slight reduction in solubility, however, fundamentally alters the properties of the tau species. Such tau aggregates are not only precipitated by conventional fixation fluids and readily visualized but, more importantly, they are no longer subject to regulation by local kinases and phosphatases: that is, the abnormal material persists in its hyperphosphorylated state and becomes completely resistant to autophagy and other endogenous cellular removal mechanisms (Kovacech et al. 2010).
Thus, it is likely that it is also primarily these minimally aggregated forms of irreversibly hyperphosphorylated tau that exert a toxic or undesirable influence on their surroundings (Kopeikina et al. 2012). To evade such effects, involved neurons convert the protein as quickly as possible into larger tau aggregates that subsequently become building blocks for neurofibrillary inclusions. Formation of the AT8-immunoreactive (AT8-ir) abnormal material continues from the outset (stage a) until the end-stage (stage VI) of AD and is not known to undergo spontaneous remission (Grundke-Iqbal et al. 1986; Tolnay and Probst 1999; Trojanowski and Lee 2000; Binder et al. 2004; Goedert et al. 2006; Mandelkow et al. 2007; Kolarova et al. 2012).
Related posts:
 Basic Organization of Non-thalamic Nuclei with Diffuse Cortical Projections
Early Presymptomatic Stages
Technical Addendum
The Pattern of Cortical Lesions in Preclinical Stages
Introduction
The Pattern of Lesions During the Transition to the Symptomatic Phase and in Fully Developed Alzheimer’s Disease
Basic Organization of Non-thalamic Nuclei with Diffuse Cortical Projections
Early Presymptomatic Stages
Technical Addendum
The Pattern of Cortical Lesions in Preclinical Stages
Introduction
The Pattern of Lesions During the Transition to the Symptomatic Phase and in Fully Developed Alzheimer’s Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


