9
Motor Disorders
APPROACH TO DIAGNOSIS
Normal motor function depends on the transmission of signals from the brain to the brainstem or spinal cord by upper motor neurons, and from there to skeletal muscle by lower motor neurons (Figure 9-1). Accordingly, motor function can be impaired by a lesion that involves this pathway anywhere along its length. Anatomic structures involved in the regulation or execution of motor activity include the pyramidal and extrapyramidal systems, the cerebellum, and the lower motor neurons in the cranial nerve nuclei of the brainstem and the anterior horns of the spinal cord.
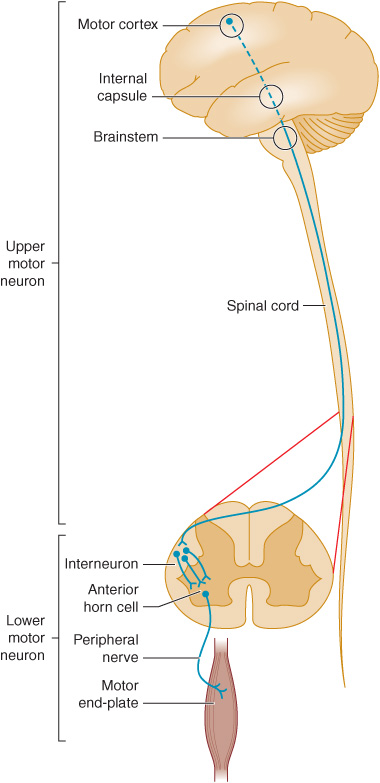
Figure 9-1. Anatomic basis of the upper motor neuron and lower motor neuron concepts.
The pyramidal system (Figure 9-2) consists of upper motor neuron fibers that descend from the cerebral cortex through the internal capsule, traverse the medullary pyramid, and then mostly decussate, to descend in the lateral corticospinal tract on the side opposite that of their origin, where they synapse on interneurons and lower motor neurons in the spinal cord.
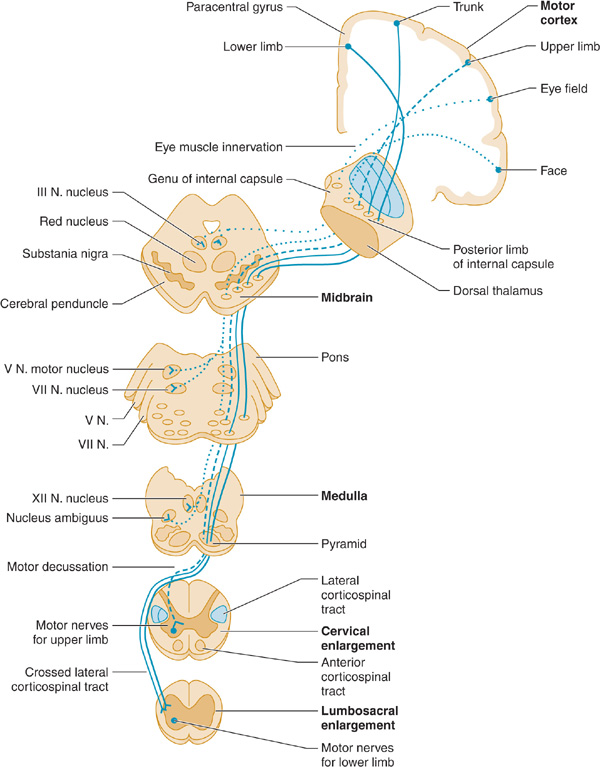
Figure 9-2. Upper motor neuron pathways. Tracts at bottom left are shown outside the cord for clarity only. (From McPhee SJ, Hammer GD: Pathophysiology of Disease: An Introduction to Clinical Medicine. 6e. New York:2009, McGraw Hill.)
All other descending influences on lower motor neurons belong to the extrapyramidal system and originate primarily in the basal ganglia and cerebellum. Disorders of the basal ganglia (Chapter 11) and cerebellum (Chapter 8) are considered separately.
The motor fibers that make up the cranial and peripheral nerves have their origin in the lower motor neurons (Figure 9-3). A disturbance of function at any point in the peripheral nervous system (anterior horn cell, nerve root, limb plexus, peripheral nerve, or neuromuscular junction) can disturb motor function, as can disease that primarily affects the muscles themselves.
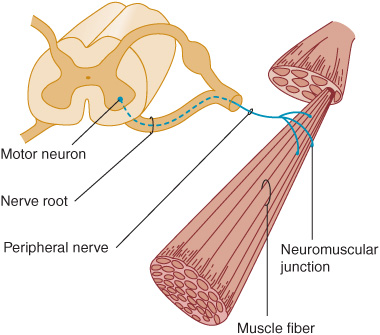
Figure 9-3. Anatomic components of the motor unit.
HISTORY
Patients with motor deficits generally complain of weakness, heaviness, stiffness, clumsiness, impaired muscular control, or difficulty in executing movements. The term weakness is sometimes used in a nonspecific way to denote fatigue or loss of energy, drive, or enthusiasm, and care must be taken to clarify what the patient means. The word is properly used to mean loss of muscle power, and it is in this sense that it is employed here.
HISTORY OF PRESENT ILLNESS
Several aspects of the present complaint must be documented.
Mode of Onset
An abrupt onset suggests a vascular disturbance, such as a stroke, or certain toxic or metabolic disturbances, whereas subacute onset over days to weeks is commonly associated with a neoplastic, infective, or inflammatory process (Table 9-1). Weakness that evolves slowly over several months or years often has a hereditary, degenerative, endocrinologic, or neoplastic basis.
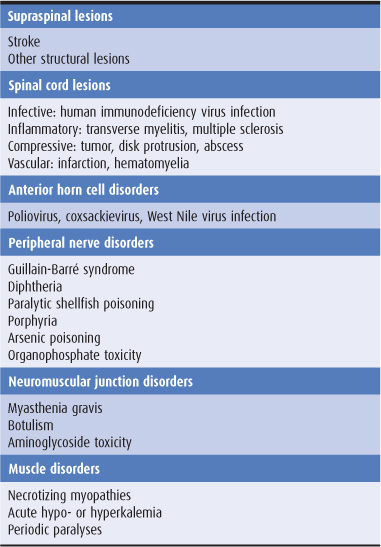
Table 9-1. Some causes of weakness of acute or subacute onset.
Course
A progressive increase in the motor deficit from its onset suggests continuing activity of the underlying process. Episodic progression suggests a vascular or inflammatory origin; a steadily progressive course is more suggestive of neoplastic disorder or such degenerative conditions as motor neuron disease. Rapid fluctuation of symptoms over short periods (eg, activity leads to fatigue and an exacerbation of weakness; rest is followed by recovery of strength) is characteristic of myasthenia gravis.
Distribution of Symptoms
The distribution of weakness and the presence of associated symptoms may indicate the approximate site of the lesion. For example, weakness in the right arm and leg may result from a lesion of the contralateral motor cortex or the corticospinal pathway at any point above the fifth cervical segment of the spinal cord. Associated right facial weakness indicates that the lesion must be above the level of the facial (VII) nerve nucleus in the brainstem, and an accompanying aphasia (Chapter 1) or visual field defect (Chapter 7) localizes it to the cerebral hemisphere.
Associated Symptoms
The presence and distribution of any sensory abnormalities are also helpful in localizing the lesion in patients with weakness. Sensory abnormalities lateralized to the same side as weakness suggest a hemispheric lesion; a cortical lesion is implied by sensory neglect or inattention, agraphesthesia (inability to identify by touch a number written on the skin), astereognosis (inability to identify by touch an object placed in the hand), abarognosis (inability to judge the weight of an object placed in the hand), or impaired two-point discrimination, when peripheral sensory function is intact. Sensory loss below a particular segmental level on the trunk suggests a spinal cord lesion, whereas distal sensory changes in the limbs favor a peripheral nerve lesion. Diseases of the anterior horn cells, neuromuscular junctions, or muscles are not accompanied by alterations in sensation.
The character of the associated symptoms may suggest the nature of the lesion at any given site in the nervous system. Thus progressive leg weakness caused by myelopathy is often preceded or accompanied by pain in the back or legs when the myelopathy is due to a compressive lesion—but not when it has a metabolic or hereditary basis.
Severity of Symptoms
An attempt must be made to evaluate the functional severity of a motor deficit by determining whether there has been any restriction of daily activities, difficulty in performing previously easy tasks, or reduction in exercise tolerance.
The nature of the functional disturbance depends on the muscles involved. Weakness of proximal muscles in the legs leads to difficulty in climbing or descending stairs or in getting up from a squatting position, whereas weakness in the arms leads to difficulty with such tasks as combing the hair. Distal weakness in the arms may lead to clumsiness, difficulty with such fine motor tasks as doing up buttons or tying shoelaces, and eventually the inability to pick up or grasp objects with the hands, so that even eating becomes difficult or impossible.
Involvement of the muscles supplied by the cranial nerves may lead to diplopia (oculomotor [III], trochlear [IV], or abducens [VI] nerve); difficulty in chewing (trigeminal [V] nerve) or sucking, blowing, or grimacing (facial [VII] nerve); or difficulty in swallowing, with nasal regurgitation and dysarthria (glossopharyngeal [IX], vagus [X], and hypoglossal [XII] nerves).
Weakness of the respiratory muscles leads to tachypnea, the use of accessory muscles of respiration, and anxiety at a stage when arterial blood gases are usually still normal. A vital capacity of less than 1 L in an adult generally calls for ventilatory support, especially if weakness is increasing.
PAST MEDICAL HISTORY
The importance of the history depends on the patient’s present complaint and the nature of any previous illnesses. For example, in a patient with known carcinoma of the lung, limb weakness may be due to metastasis or to a remote (nonmetastatic) complication of cancer. Leg weakness in a diabetic may reflect peripheral nerve, plexus, or multiple root involvement, and hand weakness in a myxedematous patient may be associated with carpal tunnel syndrome.
The history should include careful note of all drugs taken by the patient. Drugs can cause peripheral neuropathy, impair neuromuscular transmission, or lead to myopathy (Table 9-2).
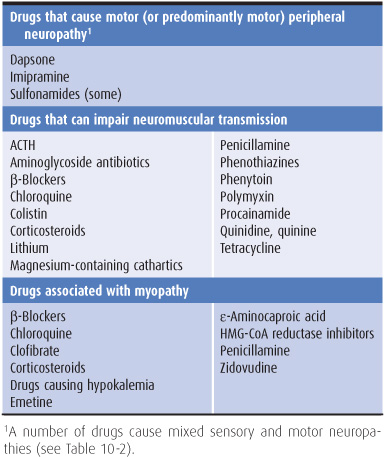
Table 9-2. Motor disorders associated with drugs.
DEVELOPMENTAL HISTORY
When symptoms develop during infancy, childhood, or early adult life, it is particularly important to obtain a full developmental history, including details of the delivery, the birth weight, the patient’s condition in the neonatal period, and the dates at which motor milestones were attained. Congenital or perinatal cerebral disease accounts for most causes of infantile diplegia (weakness of all four limbs, with the legs more severely affected than the arms).
FAMILY HISTORY
Hereditary factors may be important, and the patient’s family background therefore must be explored. Some types of myopathy, motor neuron disease, and peripheral neuropathy have a genetic basis, as do some spinocerebellar degenerations, hereditary spastic paraparesis, and certain other neurologic disorders. In some instances, it may be necessary to examine other family members to determine whether the patient’s disorder has a hereditary basis.
NEUROLOGIC EXAMINATION
MOTOR SYSTEM
In examining the motor system, a systematic approach will help to avoid overlooking important abnormalities. A sequential routine for the examination should be developed.
Muscle Appearance
1. Wasting, or muscle atrophy, suggests that weakness is due to a lesion of the lower motor neurons or the muscle itself. The distribution of wasting may help to localize the underlying disorder. Upper motor neuron disorders are not usually accompanied by muscle wasting, though muscle atrophy may occasionally occur with prolonged disuse.
2. Pseudohypertrophy of muscles occurs in certain forms of myopathy, but the apparently enlarged muscles are actually weak and flabby.
3. Fasciculations—visible irregular flickerings over the surface of the affected muscle caused by spontaneous contractions of individual motor units—suggest that weakness is due to a lower motor neuron lesion. Fasciculations are most apt to be seen in anterior horn cell disorders, but also occur in normal individuals.
4. Flexor or extensor spasms of the limbs are sometimes seen in upper motor neuron disorders as a result of impaired supraspinal control of reflex activity.
Muscle Tone
For clinical purposes, tone can be defined as the resistance of muscle to passive movement of a joint. Tone depends on the degree of muscle contraction and on the mechanical properties of muscle and connective tissue. The degree of muscle contraction depends, in turn, on the activity of anterior horn cells, which is governed by spinal and supraspinal mechanisms.
Tone is assessed by observing the position of the extremities at rest, by palpating the muscle belly, and particularly by determining the resistance to passive stretch and movement. To assess resistance to passive movement, the patient is asked to relax while each limb is examined in turn by passively taking the major joints through their full range of movement at different speeds and estimating whether the force required is more or less than normal.
Postural abnormalities may result from the increased activity of certain muscle groups caused by disturbances of reflex function, as exemplified by the typical hemiplegic posture—flexion of the upper limb and extension of the ipsilateral lower limb—of many patients who have had a stroke.
1. Hypertonia—Two types of increased tone can be distinguished.
a. Spasticity—consists of an increase in tone that affects different muscle groups to different extents. In the arms, tone is increased to a greater extent in the flexor and adductor muscles than in the extensors and abductors; in the legs, tone is increased to a greater extent in the extensor muscles than in the flexors. Moreover, the resistance of an affected muscle is not the same throughout the range of movement, but tends to be most marked when passive movement is initiated and then diminishes as the movement continues (the clasp-knife phenomenon). The increase in tone is velocity-dependent, so that passive movement at a high velocity—but not at lower velocities—may be met with increased resistance. Spasticity is caused by an upper motor neuron lesion, such as a stroke that involves the supplementary motor cortex or corticospinal tract. Spasticity may not become apparent for several days after the onset of an acute lesion, however.
b. Rigidity—consists of increased resistance to passive movement that is independent of the direction of the movement; that is, it affects agonist and antagonist muscle groups equally. The term lead-pipe rigidity is sometimes used for descriptive purposes, whereas cogwheel rigidity is used when there are superimposed ratchet-like interruptions in the passive movement, which probably relate to underlying tremor. In general, rigidity indicates extrapyramidal dysfunction and is due to a lesion of the basal ganglia (eg, Parkinson disease).
2. Hypotonia (flaccidity)—This is characterized by excessive floppiness—a reduced resistance to passive movement—so that the distal portion of the limb is easily waved to and fro when the extremity is passively shaken. In hypotonic limbs it is often possible to hyperextend the joints, and the muscle belly may look flattened and feel less firm than usual. Although hypotonia usually relates to pathologic involvement of the lower motor neuron supply to the affected muscles, it can also occur with primary muscle disorders, disruption of the sensory (afferent) limb of the reflex arc, cerebellar disease, and certain extrapyramidal disorders such as Huntington disease, as well as in the acute stage of a pyramidal lesion.
3. Paratonia—Some patients give the impression of being unable to relax and will move the limb being examined as the physician moves it, despite instructions to the contrary. In more advanced cases, there seems to be rigidity when the examiner moves the limb rapidly but normal tone when the limb is moved slowly. This phenomenon—paratonia—is particularly apt to occur in patients with frontal lobe or diffuse cerebral disease.
Muscle Power
When muscle power is to be tested, the patient is asked to resist pressure exerted by the examiner. On the basis of the history and other findings, muscles that are particularly likely to be affected are selected for initial evaluation, and other muscles are subsequently examined to determine the distribution of the weakness more fully and to shorten the list of diagnostic possibilities. For instance, if an upper motor neuron (pyramidal) lesion is suspected, the extensors and abductors of the upper extremity and the flexors of the lower extremity are tested in the most detail, because these muscles will be the most affected. Strength on the two sides is compared so that minor degrees of weakness can be recognized.
1. Upper vs. lower motor neuron lesions—Weakness can result from a disturbance in function of the upper or the lower motor neurons; the distribution of weakness is of paramount importance in distinguishing between these two possibilities. Upper motor neuron lesions (eg, stroke) lead to weakness that characteristically involves the extensors and abductors more than the flexors and adductors of the arms—and the flexors more than the extensors of the legs. Lower motor neuron lesions produce weakness of the muscles supplied by the affected neurons; the particular distribution of the weakness may point to lower motor neuron involvement at the spinal cord, nerve root, plexus, or peripheral nerve level.
2. Myopathic vs. neuropathic disorders—Weakness may also result from a primary muscle disorder (myopathy) or from a disorder of neuromuscular transmission. In patients with a motor deficit in all limbs that is not due to an upper motor neuron lesion, proximal distribution of weakness suggests a myopathic disorder, whereas predominantly distal involvement suggests a lower motor neuron disturbance.
3. Neuromuscular junction disorders—Marked variability in the severity and distribution of weakness over short periods of time (eg, over the course of a day) suggests myasthenia gravis, a disorder of neuromuscular transmission.
4. Psychogenic disorders—Apparent weakness that is not organic in nature also shows a characteristic variability; it is often more severe on formal testing than is consistent with the patient’s daily activities. Moreover, palpation of antagonist muscles commonly reveals that they contract each time the patient is asked to activate the agonist.
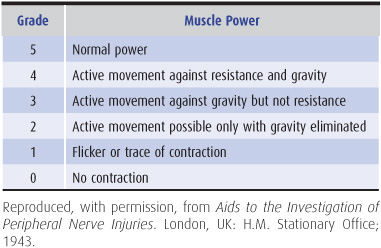
5. Muscle power grading—For practical and comparative purposes, power is best graded in the manner shown in Table 9-3.
6. Terminology for describing distribution of weakness—Monoplegia denotes paralysis or severe weakness of the muscles in one limb, and monoparesis denotes less severe weakness in one limb, although the two words are often used interchangeably. Hemiplegia or hemiparesis is weakness in both limbs (and sometimes the face) on one side of the body; paraplegia or paraparesis is weakness of both legs; and quadriplegia or quad-riparesis (also tetraplegia, tetraparesis) is weakness of all four limbs.
COORDINATION
The coordination of motor activity can be impaired by weakness, sensory disturbances, or cerebellar disease and requires careful evaluation.
Voluntary activity is observed with regard to its accuracy, velocity, range, and regularity, and the manner in which individual actions are integrated to produce a smooth complex movement.
In the finger-nose test, the patient moves the index finger to touch the tip of his or her nose and then the tip of the examiner’s index finger; the examiner can move his or her own finger about during the test to change the location of the target and should position it so that the patient’s arm must extend fully to reach it.
In the heel-knee-shin test, the recumbent patient lifts one leg off the bed, flexes it at the knee, places the heel on the other knee, and runs the heel down the shin as smoothly as possible.
Rapid alternating movement is tested by asking the patient to tap repetitively with one hand on the back of the other, to tap alternately with the palm and back of one hand on the back of the other hand or on the knee, to screw an imaginary light bulb into the ceiling with each arm in turn, and to rub the fingers of one hand in a circular polishing movement on the back of the other hand. Other tests of rapid alternating movement include tapping on the ball of the thumb with the tip of the index finger or tapping the floor as rapidly as possible with the sole while keeping the heel of the foot in place. During all these tests, the examiner looks for irregularities of rate, amplitude, and rhythm and for precision of movements. With pyramidal lesions, fine voluntary movements are performed slowly. With cerebellar lesions, the rate, rhythm, and amplitude of such movements are irregular.
If loss of sensation may be responsible for impaired coordination, the maneuver should be repeated both with eyes closed and with visual attention directed to the limb; with visual feedback the apparent weakness or incoordination will improve. In patients with cerebellar disease, the main complaint and physical finding are often of incoordination, and examination may reveal little else. Further discussion of the ataxia of cerebellar disease and the various terms used to describe aspects of it can be found in Chapter 8.
TENDON REFLEXES
Changes in the tendon (muscle stretch) reflexes may accompany disturbances in motor (or sensory) function and provide a guide to the cause of the motor deficit. The tendon is tapped with a reflex hammer to produce a sudden brisk stretch of the muscle and its contained spindles. The clinically important stretch reflexes and the nerves, roots, and spinal segments subserving them are indicated in Table 9-4. When the reflexes are tested, the limbs on each side should be placed in identical positions and the reflexes elicited in the same manner.
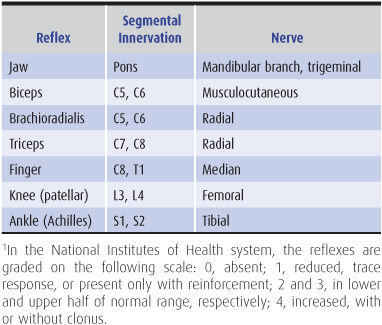
Table 9-4. Tendon (muscle stretch) reflexes.1
1. Areflexia—Apparent loss of the tendon reflexes in a patient may merely reflect a lack of clinical expertise on the part of the examiner. Performance of the Jendrassik maneuver (an attempt by the patient to pull apart the fingers of the two hands when they are hooked together) or some similar distracting action (such as making a fist with the hand that is not being tested) may elicit the reflex response when it is otherwise unobtainable. A reflex may be lost or depressed by any lesion that interrupts the structural or functional continuity of its reflex arc, as in a root lesion or peripheral neuropathy. In addition, reflexes are often depressed during the acute stage of an upper motor neuron lesion, in patients who are deeply comatose, and in patients with cerebellar disease.
2. Hyperreflexia—Increased reflexes occur with upper motor neuron lesions, but they may also occur with symmetric distribution in certain healthy subjects and in patients under emotional tension. The presence of reflex asymmetry is therefore of particular clinical significance. Clonus consists of a series of rhythmic reflex contractions of a muscle that is suddenly subjected to sustained stretch, with each beat caused by renewed stretch of the muscle during relaxation from its previous contracted state. Sustained clonus—more than three or four beats in response to sudden sustained stretch—is always pathologic and is associated with an abnormally brisk reflex. In hyperreflexic states, there may be spread of the region from which a particular reflex response can be elicited. For example, elicitation of the biceps reflex may be accompanied by reflex finger flexion, or eliciting the finger flexion reflex may cause flexion of the thumb (Hoffmann sign).
3. Reflex asymmetry—Although the intensity of reflex responses varies considerably among subjects, reflexes should be symmetric in any individual. Several general points can be made regarding reflex asymmetries.
a. Lateralized asymmetries of response—reflexes that are brisker on one side of the body than on the other—usually indicate an upper motor neuron disturbance, but sometimes reflect a lower motor neuron lesion on the side with the depressed reflexes.
b. Focal reflex deficits often relate to root, plexus, or peripheral nerve lesions. For example, unilateral depression of the ankle jerk commonly reflects an S1 radiculopathy resulting from a lumbosacral disk lesion.
c. Loss of distal tendon reflexes (especially ankle jerks), with preservation of more proximal ones, is common in polyneuropathies.
SUPERFICIAL REFLEXES
1. The polysynaptic superficial abdominal reflexes, which depend on the integrity of the T8-12 spinal cord segments, are elicited by gently stroking each quadrant of the abdominal wall with a blunt object such as a wooden stick. A normal response consists of contraction of the muscle in the quadrant stimulated, with a brief movement of the umbilicus toward the stimulus. Asymmetric loss of the response may be of diagnostic significance. The response may be depressed or lost on one side in patients with an upper motor neuron disturbance affecting that side. Segmental loss of the response may relate to local disease of the abdominal wall or its innervation, as in a radiculopathy. Bilaterally absent responses are usually of no significance, occurring in the elderly, the obese, multiparous women, and patients who have had abdominal surgery.
2. The cremasteric reflex, mediated through the L1 and L2 reflex arcs, consists of retraction of the ipsilateral testis when the inner aspect of the thigh is lightly stroked; it is lost in patients with a lesion involving these nerve roots. It is also lost in patients with contralateral upper motor neuron disturbances.
3. Stimulation of the lateral border of the foot in a normal adult leads to plantar flexion of the toes and dorsiflexion of the ankle. The Babinski response consists of dorsiflexion of the big toe and fanning of the other toes in response to stroking the lateral border of the foot, which is part of the S1 dermatome; flexion at the hip and knee may also occur. Such an extensor plantar response indicates an upper motor neuron lesion involving the contralateral motor cortex or the corticospinal tract. It can also be found bilaterally in anesthetized or comatose subjects, in patients who have just had a seizure, and in normal infants.
An extensor plantar response can also be elicited, though less reliably, by such maneuvers as pricking the dorsal surface of the big toe with a pin (Bing sign), firmly stroking down the anterior border of the tibia from knee to ankle (Oppenheim maneuver), squeezing the calf muscle (Gordon maneuver) or Achilles tendon (Schafer maneuver), flicking the little toe (Gonda maneuver), or stroking the back of the foot just below the lateral malleolus (Chaddock maneuver). In interpreting responses to these maneuvers, attention must be focused only on the direction in which the big toe first moves.
GAIT
In evaluating gait, the examiner first observes the patient walking at a comfortable pace. Attention is directed at the stance and posture; the facility with which the patient starts and stops walking and turns to either side; the length of the stride; the rhythm of walking; the presence of normally associated movements, such as swinging of the arms; and any involuntary movements (Figure 1-25).
Subtle gait disorders become apparent only when the patient is asked to run, walk on the balls of the feet or the heels, hop on either foot, or walk heel-to-toe along a straight line. Gait disorders occur in many neurologic disturbances and in other contexts that are beyond the scope of this chapter. A motor or sensory disturbance may lead to an abnormal gait whose nature depends on the site of pathologic involvement. Accordingly, the causes and clinical types of gait disturbance are best considered together.
1. Apraxic gait—Apraxic gait occurs in some patients with disturbances, usually bilateral, of frontal lobe function, such as may occur in hydrocephalus or progressive dementing disorders. There is no weakness or incoordination of the limbs, but the patient is unable to stand unsupported or to walk properly—the feet seem glued to the ground. If walking is possible at all, the gait is unsteady, uncertain, and short-stepped, with marked hesitation (“freezing”), and the legs are moved in a direction inappropriate to the center of gravity.
2. Corticospinal lesions—A corticospinal lesion, irrespective of its cause, can lead to a gait disturbance that varies in character depending on whether there is unilateral or bilateral involvement.
a. In patients with hemiparesis, the selective weakness and spasticity lead to a gait in which the affected leg must be circumducted to be advanced. The patient tilts at the waist toward the normal side and swings the affected leg outward as well as forward, thus compensating for any tendency to drag or catch the foot on the ground because of weakness in the hip and knee flexors or the ankle dorsiflexors. The arm on the affected side is usually held flexed and adducted. In mild cases, there may be no more than a tendency to drag the affected leg, so that the sole of that shoe tends to be excessively worn.
b. With severe bilateral spasticity, the legs are brought stiffly forward and adducted, often with compensatory movements of the trunk. Such a gait is commonly described as scissors-like. This gait is seen in its most extreme form in children with spastic diplegia from perinatally acquired static encephalopathy. In patients with mild spastic paraparesis, the gait is shuffling, slow, stiff, and awkward, with the feet tending to drag.
3. Frontal disorders—Some patients with frontal lobe or white matter lesions have a gait characterized by short, shuffling steps; hesitation in starting (“ignition failure”) or turning; unsteadiness; and a wide or narrow base. Sometimes referred to as marche à petit pas, this abnormality may be mistaken for a parkinsonian gait, but the wide base, preserved arm swing, absence of other signs of parkinsonism, and accompanying findings of cognitive impairment, frontal release signs, pseudobulbar palsy, pyramidal deficits, and sphincter disturbances are helpful in indicating the correct diagnosis. In patients with frontotemporal dementia, however, a parkinsonian gait and other extrapyramidal findings may be present.
4. Extrapyramidal disorders—Extrapyramidal disorders can produce characteristic gait disturbances.
a. In advanced parkinsonism, the patient is often stooped and has difficulty in beginning to walk. Indeed, the patient may need to lean farther and farther forward while walking in place in order to advance; once in motion, there may be unsteadiness in turning and difficulty in stopping. The gait itself is characterized by small strides, often taken at an increasing rate until the patient is almost running (festination), and by loss of the arm swinging that normally accompanies locomotion. At times, as when walking through a doorway, the patient may be unable to advance (“freezing”). Turning may require several small steps. In mild parkinsonism, a mildly slowed or unsteady gait, flexed posture, or reduced arm swinging may be the only abnormality found.
b. Abnormal posturing of the limbs or trunk is a feature of dystonia; it can interfere with locomotion or lead to a distorted and bizarre gait.
c. Chorea can cause an irregular, unpredictable, and unsteady gait, as the patient dips or lurches from side to side. Choreiform movements of the face and extremities are usually well in evidence.
d. Tremor that occurs primarily on standing (orthostatic tremor) may lead to an unsteady, uncertain gait, with hesitancy in commencing to walk.
5. Cerebellar disorders—In cerebellar disorders (Chapter 8), the gait may be disturbed in several ways.
a. Truncal ataxia results from involvement of midline cerebellar structures, especially the vermis. The gait is irregular, clumsy, unsteady, uncertain, and broad-based, with the patient walking with the feet wide apart for additional support. Turning and heel-to-toe walking are especially difficult. There are often few accompanying signs of a cerebellar disturbance in the limbs. Causes include midline cerebellar tumors and the cerebellar degeneration that can occur with alcoholism or hypothyroidism, as a nonmetastatic complication of cancer, and with certain hereditary disorders.
b. In extreme cases, with gross involvement of midline cerebellar structures (especially the vermis), the patient cannot stand without falling.
c. A lesion of one cerebellar hemisphere leads to an unsteady gait in which the patient consistently falls or lurches toward the affected side.
6. Vestibular disorders—With unilateral vestibular dysfunction, the patient is unsteady, veering to the affected side. If both sides are affected, the gait becomes especially unsteady in the dark, when visual input is reduced.
7. Impaired sensation—Impaired sensation, especially disturbed proprioception, also leads to an unsteady gait that is aggravated by walking in the dark or with the eyes closed, as visual input cannot then compensate for the sensory loss. Because of their defective position sense, many patients lift their feet higher than necessary when walking, producing a steppage gait. Causes include tabes dorsalis, sensory neuropathies, vitamin B12 deficiency, and certain hereditary disorders (Chapter 10).
8. Anterior horn cell, peripheral motor nerve, or skeletal muscle disorders—These disorders lead to gait disturbances if the muscles involved in locomotion are affected. Weakness of the anterior tibial muscles leads to foot drop; to avoid catching or scuffing the foot on the ground, the patient must lift the affected leg higher than the other, in a steppage gait resembling that due to sensory disorders. Weakness of the calf muscles leads to an inability to walk on the balls of the feet. Weakness of the trunk and girdle muscles, such as occurs in muscular dystrophy, other myopathic disorders, and Kugelberg-Welander syndrome, leads to a waddling gait because the pelvis tends to slump toward the non–weight-bearing side.
9. Unsteady or cautious gait in the elderly—
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


