35 Myopathy
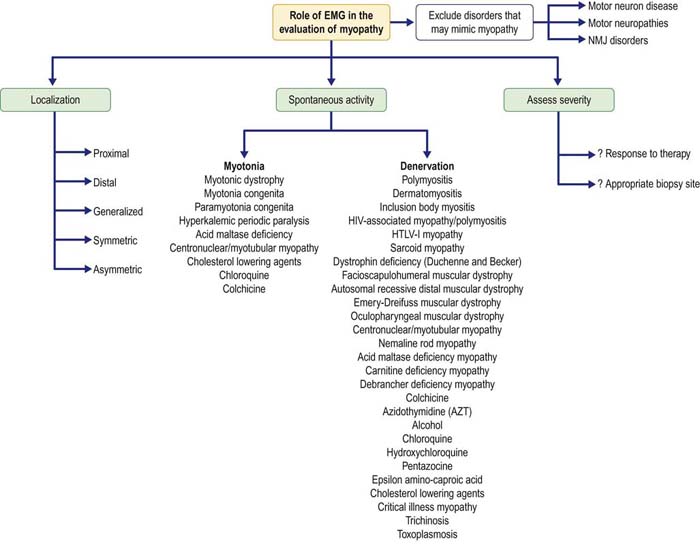
In the evaluation of patients with suspected myopathy, molecular genetics has supplanted the need for electrodiagnostic (EDX) studies or muscle biopsy in many patients with inherited conditions. Moreover, in patients with suspected myopathy and no evidence of an inherited condition, a muscle biopsy ultimately will be required for definitive diagnosis, regardless of EDX studies. Despite these facts, EDX studies, especially the needle electromyography (EMG) examination, continue to play an important role in the evaluation of patients with suspected myopathy (Figure 35–1). EMG can often confirm the presence of a myopathy, as well as add diagnostic information if certain types of spontaneous activity are present. For example, fibrillation potentials and positive sharp waves in a myopathy suggest the possibility of inflammation or necrosis, whereas myotonic discharges suggest one of the myotonic muscle or periodic paralysis disorders (see Chapter 36), acid maltase deficiency, myotubular myopathy, or certain toxic myopathies. Additionally, EMG may be helpful in suggesting alternate diagnoses that can mimic myopathy clinically.
Clinical
Myopathy associated with periodic paralysis occurs in the setting of hypokalemic and hyperkalemic periodic paralysis (see Chapter 36). Patients develop proximal weakness in the fifth or sixth decade. Even those patients with hypokalemic periodic paralysis who have never experienced episodic weakness, a common scenario in affected females, invariably will develop a proximal vacuolar myopathy in adulthood.
Electrophysiologic Evaluation
Nerve Conduction Studies
Routine nerve conduction studies should always be done in patients with suspected myopathy (Box 35–1). Sensory nerve conduction studies are always normal, unless there is a coexistent neuropathy. Because most myopathies preferentially affect proximal muscles and routine motor nerve conduction studies record distal muscles, motor nerve conduction studies are also usually normal. If the myopathy is severe enough to affect distal and proximal muscles or is one of the rare myopathies that preferentially affects distal muscles, motor studies may show decreased compound muscle action potential (CMAP) amplitudes with normal latencies and conduction velocities.
Box 35–1
Recommended Nerve Conduction Study Protocol for Myopathy
1. At least one motor and one sensory conduction study and corresponding F responses from the upper extremity (e.g., median motor and sensory, median F responses)
2. At least one motor and one sensory conduction study and corresponding F responses from the lower extremity (e.g., tibial motor and sural sensory, tibial F responses)
• If the compound muscle action potential (CMAP) amplitudes are decreased or borderline, exercise the muscle maximally for 10 seconds, then repeat a single supramaximal distal stimulation, looking for a significant CMAP increment (>100% of baseline), suggestive of the diagnosis of Lambert–Eaton myasthenic syndrome.
• If there is a clinical history of fatigability, repetitive nerve stimulation (3 Hz) of one distal muscle (e.g., ulnar nerve recording abductor digiti minimi) and one proximal muscle (e.g., the spinal accessory nerve recording the upper trapezius) should be performed. If a significant decrement is found with 3 Hz repetitive nerve stimulation of any muscle, then proceed with further testing, looking for a disorder of the neuromuscular junction (see Chapter 34, Box 34–2).
The major reason nerve conduction studies must be performed is to exclude other motor disorders that may mimic myopathy (Box 35–2). Other than myopathy, pure motor disorders include motor neuron disease, rare cases of demyelinating polyneuropathy, and NMJ disorders. The nerve conduction studies in motor neuron disease and myopathies that affect distal muscles may be very similar. Differentiation is made based on the associated clinical features and needle EMG findings. Nerve conduction studies can easily differentiate demyelinating polyneuropathy from myopathy by the presence of conduction block or temporal dispersion, marked slowing of distal latencies and conduction velocity, or a combination of these findings.
Box 35–2
Disorders that May Mimic Myopathy
Neuromuscular junction disorders
Central nervous system lesions
Bilateral middle cerebral artery–anterior cerebral artery watershed strokes
Disorders of the NMJ present more of a challenge. NMJ disorders may present with proximal muscle weakness similar to myopathies. Postsynaptic disorders (e.g., MG) typically have normal CMAP amplitudes at rest. To demonstrate the NMJ abnormality, slow (3 Hz), repetitive nerve stimulation is required to demonstrate a decrement (see Chapter 34). Presynaptic disorders (e.g., LEMS) have a more characteristic nerve conduction pattern: CMAP amplitudes are low at rest with normal latencies and conduction velocities. Brief exercise (10 seconds) characteristically results in a marked increment of CMAP amplitude (typically >100% of baseline).
Electromyographic Approach
For the patient with suspected myopathy, the needle EMG examination must be individualized based on the distribution of the patient’s symptoms (Box 35–3). Overall, examining distal and proximal muscles in both the upper and lower extremities is indicated. Sampling the paraspinal muscles (the most proximal muscles) often is very useful. As most myopathies affect proximal muscles, the yield of finding abnormalities increases as progressively more proximal muscles are sampled. In adult-onset acid maltase deficiency myopathy, for instance, prominent changes may be seen only in the paraspinal muscles.
Box 35–3
Recommended Electromyographic Approach to Myopathy
1. At least two distal and two proximal muscles in the lower extremity (e.g., tibialis anterior, gastrocnemius, vastus lateralis, iliacus)
2. At least two distal and two proximal muscles in the upper extremity (e.g., first dorsal interosseous, extensor indicis proprius, biceps brachii, medial deltoid)
• Always try to study weak muscles. The number and distribution of muscles studied depend on the pattern of weakness.
• Try to study muscles that can easily be biopsied on the contralateral side (deltoid, biceps, vastus lateralis, gastrocnemius).
• If the motor unit action potential (MUAP) parameters are indeterminate, consider the following:
Spontaneous Activity in Myopathies
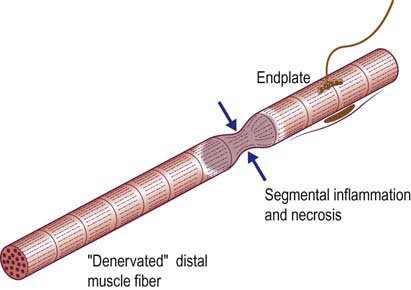
Fibrillation potentials and positive sharp waves usually are associated with neuropathic disorders (i.e., neuropathy, radiculopathy, motor neuron disease). However, denervating potentials occur frequently in many myopathic disorders. They are thought to most likely occur as a consequence of segmental inflammation or necrosis of muscle fibers, separating a distal, healthy portion of the muscle fiber from the part attached to the endplate (Figure 35–2). Infarction of small intramuscular nerve twigs by surrounding interstitial inflammation also is speculated to be a possible cause of denervation in inflammatory myopathies. Although the presence of denervating potentials in a patient with myopathy often suggests the diagnosis of an inflammatory myopathy, denervating potentials can occur in a variety of myopathies (Box 35–4). In chronic myopathies, complex repetitive discharges may also be seen.