INTRODUCTION
Pediatric neurologists treat many children with brain injuries that originate in the perinatal period and cause mental retardation, cerebral palsy, and/or seizures. This chapter will focus on some of the most common disease processes affecting neonates.
PERIVENTRICULAR-INTRAVENTRICULAR HEMORRHAGE
EPIDEMIOLOGY
Premature, low birth weight neonates are especially at risk for periventricular-intraventricular hemorrhage, and the incidence and severity of intraventricular hemorrhage (IVH) increases with earlier prematurity and lower birth weight. The prevalence of periventricular IVH in newborns weighing less than 1,500 g has declined, falling from approximately 40% in 1980 to 20% since the late 1990s. Corticosteroid therapy prenatally and surfactant use after birth have contributed to the decline of IVH. However, with the increased survival rate of extremely low birth weight infants, there are more infants at highest risk, with a risk of 45% in infants less than 1,000 g. In a population-based study in Switzerland, IVH decreased by 3.5% with each added week of gestation.
Additionally, severe IVH increases with decreasing gestational age. In a large, multicenter registry of infants of very low gestational age (22 to 28 weeks) and birth weight (401 to 1,500 g), born between 2003 and 2007 in the United States, 16% overall had severe IVH, and the prevalence increased with decreasing gestational age, from 38% at 22 weeks to 7% at 28 weeks.
PATHOBIOLOGY
In premature neonates, the vascular germinal plate near the foramen of Monro, in the region of the caudate, is friable and susceptible to hemorrhage. The risk for hemorrhage is further exacerbated by disturbances in cerebral blood flow, either through hypotension and lower cerebral perfusion, possibly lowered further by positive pressure ventilation through endotracheal intubation, and/or through hypoperfusion-reperfusion coupled with immaturity of intrinsic cerebral vasoreactivity and autoregulatory mechanisms. Other factors potentially increasing risk of IVH during prematurity are metabolic (hypoglycemia, hypernatremia), hematologic (anemia, thrombocytopenia), and immunologic (chorioamnionitis, immaturity of antioxidant responses, or other immune responses).
The Papile grading system, with some modification, is the most commonly used scale to describe the severity of the IVH (
Fig. 132.1). The IVH may remain in the matrix area (grade I), but 50% can rupture into the lateral ventricles (grade II), which can result in ventricular enlargement or distension (grade III). Grade IV, now termed
periventricular hemorrhagic infarction (PVHI), refers to extensive IVH with parenchymal involvement, likely due to terminal venous occlusion, venous infarction, and subsequent secondary hemorrhage.
Late in the course after IVH (1 to 3 weeks after the initial hemorrhage), especially with more severe IVH, there can be posthemorrhagic hydrocephalus (PHH), or progressive ventricular dilatation (PVD), thought to result from impaired reabsorption of cerebrospinal fluid (CSF) because of inflammation of the subarachnoid villi. Usually, impaired reabsorption results in a communicating hydrocephalus, although there can also be a noncommunicating hydrocephalus due to localized obstruction or scarring.
CLINICAL MANIFESTATIONS
Most IVH in premature infants occurs within the first 5 postnatal days. Late IVH is associated with low cerebral blood flow, usually from systemic processes.
The clinical presentation of IVH varies depending on severity. Grade I IVH is usually asymptomatic. Many grade II and some III hemorrhages are clinically silent, but there may be a saltatory course evolving over hours to days, characterized by nonspecific findings of altered level of consciousness, hypotonia, eye movement changes, subtle changes in movement, decreased spontaneous movements, and disturbed respiratory function. Some grade III hemorrhages produce hydrocephalic symptoms of varying severity, and if seen in combination with PVHI, there may be signs of severe apnea, bradycardia, extensor posturing, and opisthotonus. There also may be flaccid weakness, cranial nerve abnormalities including fixed pupils to light, most likely leading to unresponsiveness and death within hours. There may be clonic limb movements, which some clinicians have labeled as seizures, although there is usually little electroencephalogram (EEG) correlation. Most infants who die have other lesions, such as periventricular leukomalacia (see the section “Periventricular Leukomalacia”), brain stem necrosis, or cerebellar necrosis.
HYPOXIC-ISCHEMIC ENCEPHALOPATHY
EPIDEMIOLOGY
In developed countries, birth asphyxia affects 3 to 5 newborns per 1,000 live births and causes moderate to severe hypoxic-ischemic encephalopathy (HIE) in 0.5 to 1 per 1,000 live births. Globally, 10% to 60% of neonates with HIE die, and 25% have long-term neurodevelopmental difficulties.
PATHOBIOLOGY
HIE is most commonly associated with severe maternal hypotension, uterine rupture, placental abruption, and placental or umbilical cord dysfunction. In addition, there may be associated hypoxic cardiopulmonary, hepatic, or renal injury.
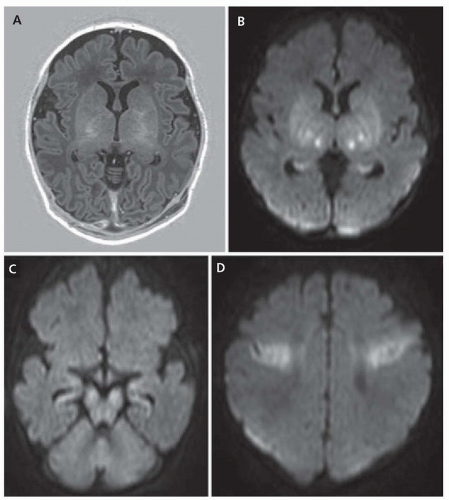
Brain pathology depends on the level of maturity at the time of the insult, as well as the duration and location of the insult. In infants injured younger than 32 weeks’ gestation, periventricular leukomalacia (PVL) is commonly seen because of the vulnerability of late oligodendroglial precursors. After 36 weeks’ gestation, hypoxic lesions are frequently located in the cerebral gray matter, deep gray nuclei, brain stem, and Purkinje cells of the cerebellum.
On a cellular and molecular level, hypoxia-ischemia acutely causes neuronal loss in the cerebral sulci, often with edema and infarction. Chronically, there is neuronal loss and astrocytosis, with possible atrophy, cystic encephalomalacia, ulegyria, status marmoratus, and cerebellar atrophy. Energy failure from hypoxia stems from loss of mitochondrial function, which can be confirmed by measuring increased lactic acid levels in the CSF or by magnetic resonance spectroscopy (MRS). There is accompanying membrane depolarization and an increase in neurotransmitter release, as well as excessive activation of glutamate receptors. Resultant is excitotoxic cellular injury, leading to an increase in intracellular calcium that leads to a cascade of pathologic pathways leading to secondary cell death. In addition, there is oxidative stress, which produces reactive oxygen species and subsequent reactive nitrogen radicals, also contributing to cell toxicity. After the anoxic insult, many cells undergo reoxygenation and reperfusion. Reperfusion contributes further to oxidative stress by producing more reactive oxygen species and further cellular injury.
CLINICAL MANIFESTATIONS
HIE initially presents with low Apgar scores, which translate into bradycardia, depressed respiration, diminished responsiveness, and decreased muscle tone (
Table 132.2). One of three clinical patterns then evolves: (1) mild: awake, irritable, hyperalert, with jitteriness, dilated pupils, increased deep tendon reflexes, and normal muscle tone; (2) moderate: lethargy with depressed deep tendon reflexes, frequently with seizures; and (3) severe: apathy, with severe hypotonia, poorly reactive pupils, an absent Moro reflex, depression or coma, and seizures. There is usually clinical improvement with the mild and moderate cases as the newborns become more alert, improve their tone, and are able to feed. Seizures or therapy for seizures may delay recovery. Newborns with severe encephalopathy will usually become stuporous; develop frequent seizures, respiratory depression, or brain stem abnormalities; and may show signs of decerebration. They become unresponsive and lose sucking reflexes and the Moro response, and even with vigorous supportive and anticonvulsant therapy, 20% to 30% of these newborns die. If the neonate survives, seizures usually stop after 48 to 72 hours, and there is usually some recovery.



 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access






