Chapter 49B Neurological Complications of Systemic Disease
Children
Cardiac Disorders and the Nervous System
Congenital Heart Disease
Cerebral Dysgenesis and Malformations
Cerebral dysgenesis is a consideration to explain neurological symptoms in children with CHD. Autopsy studies reveal a 10% to 29% prevalence of cerebral malformations. Patients with hypoplastic left heart syndrome are at a higher risk for associated brain dysgenesis. In one series of 41 patients with hypoplastic left heart syndrome, 29% had associated brain malformations of variable severity; 27% had microcephaly, 21% had immature cortical mantle, and the remainder had other malformations, one with holoprosencephaly. Other reports include agenesis of the corpus callosum, Dandy-Walker syndrome, and aqueductal stenosis. Lutterman and colleagues (1998) described the association of moyamoya disease and structural CHD, including ventricular septal defect (VSD), aortic and mitral valve stenosis, and tetralogy of Fallot.
Chromosomal and Genetic Disorders
The combination of CHD and neurological disorders, mainly developmental delay, are sometimes manifestations of genetic conditions combining both cardiac and central nervous system (CNS) involvement. Such conditions include trisomy 21, trisomy 13, trisomy 18, Williams syndrome, DiGeorge syndrome, and velocardiofacial syndrome. Chromosomal microarray analysis, also known as array-based comparative genomic hybridization (CGH), has recently become an extremely valuable diagnostic tool, allowing the detection of subtle genomic imbalances undetected by conventional chromosome analysis. Lu et al. (2008) studied 101 patients with CHD with or without other malformations such as cleft palate, club foot, and polydactyly, and array-based CGH detected significant abnormalities in 21.8% of patients. Richards et al. (2008) also found that children with CHD and other anomalies, specifically neurological problems, had a higher incidence of cryptic chromosomal abnormalities detected by CGH, which were not detected by conventional karyotyping. The authors advocated screening patients with CHD and neurological abnormalities such as developmental delay with chromosomal microarray analysis.
Neurological Complications Unrelated to Intervention and Cardiac Surgery
Infective Endocarditis
The implementation of subacute endocarditis prophylaxis before dental and surgical procedures in patients with CHD has greatly reduced the incidence of bacterial endocarditis. Approximately one-third of cases of infective endocarditis are associated with neurological complications. These include cerebral embolization, usually in the middle cerebral artery territory, and meningitis, brain abscess, and seizures (Fig. 49B.1). Cerebral mycotic aneurysms complicate 1.2% to 5% of cases of infective endocarditis and carry a high mortality rate of 60%. The risk of hemorrhagic transformation of septic infarctions is high and associated with a mortality rate of 80% to 90%.
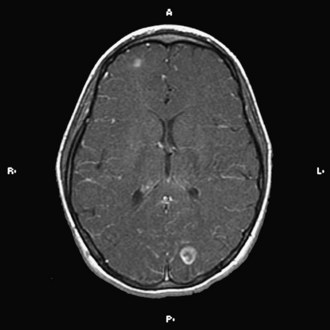
Neurological Complications of Intervention and Cardiac Surgery
Fallon and associates (1995) reviewed data for 523 cardiac surgery patients and found neurological events or deficits in 31 patients in the immediate postoperative period. Seizures occurred in 16, pyramidal signs (hemiparesis-quadriparesis) in 11, extrapyramidal signs in 8, and neuro-ophthalmic deficits (gaze palsies, visual field defects) in 6. Six patients were unconscious, and four demonstrated miscellaneous neurological changes such as development of Horner syndrome secondary to brachial plexus injury, vocal cord palsy, isolated bulbar palsy, and transient ischemic episodes. A period of low perfusion pressure, either intraoperatively or postoperatively, was present in more patients who had an adverse neurological event than in those who were normal. The highest frequency of adverse neurological events was in the cardiac diagnostic group of arch anomaly. The likely pathogenesis of CNS injury is microembolization and ischemia during bypass or development of intracranial hemorrhage (Du Plessis et al., 1999). Corrective surgery for coarctation of the aorta is especially associated with CVAs.
A postoperative encephalopathy characterized by choreoathetosis and developmental delay is a well-defined complication after cardiac surgery in children, but not after cardiac surgery in adults. The incidence has dropped from 18% to 0.6% in recent reports (du Plessis et al., 2002). A mild transitory form can follow cardiac surgery in infants. The severe form occurs in children who undergo such surgery after infancy. In the severe postpump choreoathetosis, the early mortality rate approaches 40%. Most of the patients have residual involuntary movements and severe long-term neurological disturbances years later. The mild form is associated with cognitive and behavioral disturbances despite complete resolution of choreoathetosis. The mechanisms underlying pathogenesis remain unclear, with the usual proposed explanations being deep hypothermia and intraoperative hypoxic injury (Wessel et al., 1995). Brain imaging in these cases usually reveals nonspecific changes such as cerebral atrophy. Neuropathological data are limited; however, the external globus pallidus is the most consistent locus of injury, with evidence of gliosis, neuronal loss, nerve fiber degeneration, and capillary proliferation (Kupsky et al., 1995).
Open heart surgery is associated with several risk factors for stroke (Fig. 49B.2). The risks include altered intravascular endothelial surfaces, thrombus formation facilitated by the use of prosthetic devices, gaseous emboli originating from the cardiopulmonary bypass, global hypoperfusion, inflammatory cascades and microvascular inflammatory changes, and occurrence of prothrombotic state during surgery, owing to consumptive coagulopathy and decreased protein C and antithrombin levels.
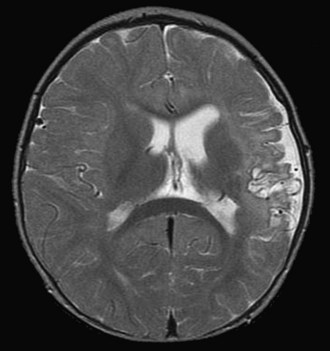
Spinal cord injury occurs especially after aortic coarctation repair. Peripheral neuromuscular complications include plexopathies (mostly brachial), pressure palsies (peroneal and ulnar nerves), myopathy, “critical care neuropathy,” and polyneuropathy developing after withdrawal of neuromuscular blocking agents. Dittrich et al. (2003) reviewed data for 90 patients younger than 1 year of age who underwent cardiac surgery. These patients had no brain anomalies or syndromes associated with delayed mental development, but 32% had evidence of psychomotor impairment. Neurological sequelae were more frequent after palliative surgery than after corrective surgery.
Cardiac Transplantation
Perez-Miralles and associates (2005) reported neurological complications after cardiac transplantation in 13.7% of patients. Other studies, however, have reported an incidence of 50% to 70%, mostly in the perioperative period.
In the series of Cemillan and colleagues (2004), 48% of transplant recipients suffered neurological complications such as encephalopathy (16.6%), seizures (13.6%), neuromuscular disorders (10.6%), headaches (10.6%), CVA (10.1%), psychiatric problems (2.2%), and CNS infections (2.2%). Signs and symptoms of cyclosporine toxicity include tremor, seizures, and encephalopathy. Risk factors for encephalopathy were renal and hepatic failure and hemodynamic instability. Risk factors for stroke were the presence of systemic hypoperfusion, arrhythmias, coagulopathies, and hypertension.
Connective Tissue Diseases and Vasculitides
Polyarteritis Nodosa
Neurological manifestations can develop in 50% to 70% of children. Mononeuritis multiplex, a characteristic feature of the disease in adults, is much less frequent in children, whereas CNS manifestations are more frequent in children than adults. Focal neurological deficits secondary to ischemia, infarction, and hemorrhage are common. The signs and symptoms include unilateral blindness, visual field defect, seizures, headache, encephalopathy, cognitive decline, cranial neuropathies, and aseptic meningitis. In the brain, changes are mainly seen in the small meningeal arteries (Nadeau et al., 2002).
Corticosteroid therapy improves life expectancy and decreases the incidence of hypertension and renal complications. In severe cases, lack of response to steroids is an indication for use of oral or intravenous-pulse cyclophosphamide. Plasmapheresis has not improved survival. Methotrexate, azathioprine, mycophenolate mofetil, intravenous immunoglobulin (IVIG) and more recently tumor necrosis factor (TNF) inhibitors (infliximab) and anti-CD20 monoclonal antibodies (rituximab) have been used successfully in children (Gedalia et al., 2009).
Kawasaki Disease
The most common neurological manifestations consist of extreme irritability, probably caused by aseptic meningitis, headaches, and encephalopathy. Cerebral infarction, seizures, polyneuropathy, myositis, cranial neuropathies, and retinal vasculitis are rare complications. Muneuchi and associates (2006) described a single patient with a silent right cerebellar infarct and suggested the need to consider the possibility of brain lesions in all children with Kawasaki disease with or without neurological symptoms.
Juvenile Rheumatoid Arthritis
The neurological complications of the systemic form include acute encephalopathy, which can be lethal as a result of the macrophage activation syndrome (Ueno et al., 2002). The cause of this syndrome is disruption of the macrophage-lymphocyte interaction, causing uncontrolled proliferation of highly activated macrophages and T lymphocytes, with consequent sepsis-like symptoms often resulting in multiple organ failure. High-grade fever, hepatosplenomegaly, pancytopenia, consumption coagulopathy, and low erythrocyte sedimentation rate are other features. Treatment is with high-dose steroids and cyclosporine (Stabile et al., 2006).
Systemic Lupus Erythematosus
CNS involvement occurs in 30% to 60% of children with SLE during the course of their illness. Neuropsychiatric abnormalities occur in as many as 95% of patients. Patients with CNS involvement usually have a more severe clinical course. The prevalence of recurrent headaches is 71%, migraine 36%, cognitive disorders 55%, isolated seizures 47%, epilepsy 15%, acute confusional state 35%, dysesthesia or paresthesia 14%, transient ischemic attacks (TIAs) 12%, and CVA 8%. Chorea and myositis are rare (Ghosh et al., 2005). Parkinsonism has been reported in three patients (Kwong et al., 2000). Corticosteroid-related myopathy can complicate the course of the disease. Ophthalmoplegia, diplopia, sudden blindness, or ptosis can occur, and findings of papilledema, optic neuritis, retinal hemorrhage, and vasculitis (“cotton wool spots”) have been described. Possible neurological complications of SLE include ataxia, vertigo, sensorineural hearing loss, aseptic meningitis, transverse myelopathy, and peripheral neuropathy with predominantly sensory deficits. The most common clinical manifestations of neuropsychiatric lupus are depression, memory problems, emotional lability, trouble with concentration, and psychosis. Psychiatric assessment is important in the evaluation of children with SLE. Muscal et al. (2010) and others have described pediatric lupus patients presenting with seizures, altered mental status, and MRI findings suggestive of posterior reversible encephalopathy syndrome (PRES).
Vasculitis in SLE is rare and affects small arterioles and venules. Perivasculitis is more common. CVAs occur mainly in patients with hypertension or severe renal and cardiac disease (Fig. 49B.3) and have been associated with positive results on serological tests for syphilis and the presence of lupus anticoagulant (LA). Gattorno and colleagues (1995) found that 79% of pediatric patients with SLE had anticardiolipin antibodies (aCLs), and 42% had LA. These patients were at high risk for the development of deep vein thrombosis and other antiphospholipid antibody (APA)-related pathology. A statistically significant correlation also has been found between APA and neurological manifestations such as vascular events, seizures, and psychosis. Other antibody systems such as antiribosomal P antibodies, antineuronal antibodies, or lymphocytotoxic antibodies also may be associated with an increased risk for neurological involvement. The pathogenesis of neuropsychiatric lupus is likely to be multifactorial, including autoantibody production, microangiopathy, intrathecal production of proinflammatory cytokines, and premature atherosclerosis. It is possible that the autoantibodies associated with neuropsychiatric lupus may require a disrupted blood-brain barrier to exert their effect (Nishimura et al., 2008).
< div class='tao-gold-member'>





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


