Chapter 20 Perinatal Metabolic Encephalopathies
Introduction
Box 20-1 Predominant Presenting Clinical Features in Neonatal Genetic Metabolic Disorders


























A brief synopsis of each condition is provided, with emphasis on distinctive clinical features, diagnostic approach, and management. Comprehensive discussion of the molecular biology, pathophysiology, and genetics of these disorders may be found in several excellent reviews [Scriver et al., 2001; Hoffmann et al., 2010].
General Approach
Metabolic disease should be considered in the differential diagnosis of any newborn demonstrating symptoms or signs of encephalopathy, which are summarized in Table 20-1. While none of these abnormalities is specific to metabolic disease, the temporal pattern and specific constellation of symptoms taken together often suggest a metabolic disease. Encephalopathies caused by acute correctable metabolic perturbations, such as in hypoglycemic encephalopathy, typically follow a monophasic course that resolves in proportion to the timeliness of correction of the metabolic perturbation. The tempo of genetically determined metabolic diseases is more variable than acquired acute injury such as hypoxia or trauma. It often has a delayed or subacute presentation with unexplained disturbances in state of arousal or neonatal behaviors, such as feeding or visual attentiveness, or with the emergence of myoclonic seizures or movement disorder. Chronic encephalopathies that initially manifest in the newborn period are increasingly being linked to determine metabolic disease genetically. These disorders may have few or no features that distinguish them as metabolic, presenting with nonspecific signs such as hypotonia, depressed neonatal behaviors, or poor feeding. Nervous system manifestations of metabolic disease are generalized in nature, with depression of consciousness or irritability. Motor abnormalities and brainstem dysfunction parallel the severity of disturbed consciousness in a nonlocalizing manner. Suspicion of an inborn error of metabolism should be elevated by any of the following patterns:
Table 20-1 Neurologic Signs and Symptoms of Neonatal Metabolic Disease
| Neurologic Examination Category | Finding in Affected Infant with Metabolic Disease |
|---|---|
| Alterations in consciousness Mild to moderate Severe | Excessively irritable, or somnolent but awakens with light stimulation Unarousable, but withdraws purposefully Comatose, responds to pain only by posturing, or not at all |
| Brainstem dysfunction | Pupillary abnormalities Oculomotor deficits Depressed or disordered suck and swallow function Disordered control of breathing (hyperpnea, apnea) High-pitched (“cerebral”) cry |
| Cortical special senses | Absent arousal to sound, poor or absent visual fixation |
| Tone and movement abnormalities | Generalized hypotonia or hypertonia Nonepileptic myoclonus Dystonia, rigidity, opisthotonus, oculogyric crises |
| Reflex abnormalities | Exaggerated deep tendon reflexes Disturbed neonatal reflex behaviors (e.g., Moro, grasp) |
| Seizures | Myoclonic, clonic, tonic, subtle, apnea |
| Neuromuscular abnormalities | Diffusely absent tendon reflexes, bilateral symmetric facial or limb weakness, respiratory insufficiency |
| Autonomic dysfunction | Temperature instability, gastrointestinal dysmotility |
Diagnosis is further delineated by appropriate staged laboratory studies, as shown in Table 20-2. The aim of the initial evaluation is to narrow down the differential diagnosis and simultaneously treat potentially life-threatening or handicapping conditions as quickly as possible. The results of “first-stage” studies will reveal abnormalities requiring immediate correction, such as hypoglycemia, hyponatremia, hypocalcemia, or hypoxia. They will further define other organ involvement, such as renal, cardiac, or hepatocellular dysfunction. A constellation of abnormalities on the initial screening testing may suggest a particular inborn error of metabolism (Figure 20-1). If the results of first-stage assessment, combined with clinical history, examination, and neuroimaging, do not provide a specific diagnosis, they should provide the rationale for choosing appropriate “second-stage” studies. These are aimed at identifying the category of metabolic defect, and in some cases provide a probable specific diagnosis that can be verified in “third-stage” studies, in which DNA or tissue sampling provides material for enzyme quantification or genetic analysis. A careful family history should be obtained to ascertain consanguinity, known inherited metabolic disease, excess fetal loss, or infant demise in a sibling under similar circumstances.
Table 20-2 Laboratory Evaluation of the Infant with Suspected Metabolic Encephalopathy
| First Stage Survey and Screening | Second Stage Identify Category of Metabolic Defect | Third Stage Verify Enzyme or Gene Defect |
|---|---|---|
| Blood Glucose Electrolytes Mg++, Ca++, PO4 BUN, creatinine Liver enzymes CBC, platelets, PT, PTT Arterial blood gas Ammonia Lactate, pyruvate | Blood Quantitative plasma amino acids Very long chain fatty acids Acyl carnitine profile Isoelectric transferrin point Copper, ceruloplasmin levels Cholesterol Uric acid | Tissue biopsy (muscle, liver) for enzyme assay Skin fibroblast culture for enzyme assay DNA studies and specific gene mutation probes Family studies |
| Urine Ketone bodies, reducing substances Protein, blood Cells | Urine Quantitative amino acids Organic acids Galactose Sulfites Xanthine, hypoxanthine Uric acid Ribosides | |
| CSF Glucose, protein Cell count Microbiology | CSF Quantitative amino acids Neurotransmitters – Biopterin metabolites Lactate, pyruvate |
BUN, blood urea nitrogen; CBC, complete blood count; CSF, cerebrospinal fluid; PT, prothrombin time; PTT, partial thromboplastin time.
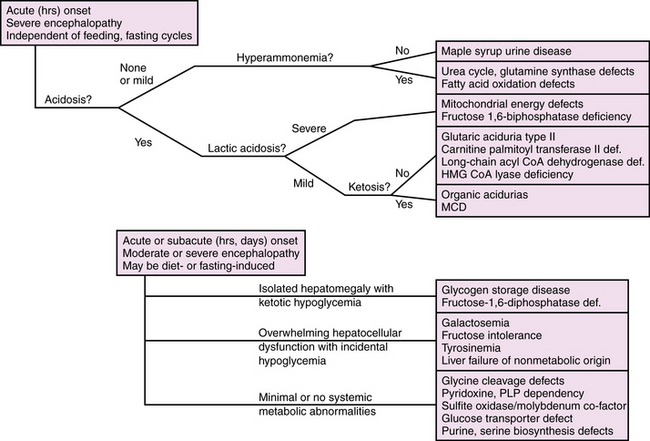
Fig. 20-1 Algorithm for evaluation of suspected metabolic encephalopathy in neonates.
MCD, malonyl CoA decarboxylase deficiency; PLP, pyridoxine and pyridoxal 5′-phosphate dependency.
Correctable Disturbances of Glucose and Salt Balance
Hypoglycemia
Hypoglycemia is common among critically ill newborns and is a frequent manifestation of inborn errors of metabolism. The definition of hypoglycemia is controversial. Blood glucose values reflect a dynamic balance between substrate availability and utilization, and in healthy newborns vary across a broad range, typically >2.2 μM (36 mg/dL), but reported as low as 1.3 μM (23 mg/dL) in healthy breastfed infants in the first few days of life. The risk of neurologic injury from hypoglycemia depends on multiple factors, including gestational age, availability of alternate substrates, metabolic rate, maturity and efficiency of glucose transport systems, status of oxidative metabolism, and differences in regional brain metabolic activity. As such, clinical factors such as coexistent hypoxic-ischemic injury, seizures, and the presence of hyperinsulinism strongly influence the occurrence and nature of hypoglycemia-related neuronal injury. These observations have led a number of investigators to propose an “operational” definition, whereby threshold blood glucose value, for example, 2.5 mmol/L (46 mg/dL), prompts treatment and further evaluation. This threshold may be modified up or down in individual cases, depending on clinical symptoms and factors likely to affect brain substrate demand, such as seizures or hypoxia, and alternative substrate availability, such as lactate or ketones or the effects of hyperinsulinism. Presently, there is insufficient evidence to define specific threshold values as predictive of neurologic injury in all clinical settings, and further research is needed on this point [Hay et al., 2009].
Disorders leading to neonatal hypoglycemia may be grouped into four broad categories: maladaptation to extrauterine life, hyperinsulinism, increased glucose consumption due to prior or intercurrent illness, and inborn errors of metabolism (Table 20-3). Maladaptation to extrauterine life affects premature infants and small-for-gestational-age infants due to low stores of fat, protein, and glycogen; inefficient ketogenesis and gluconeogenesis; or diminished tolerance of feedings. Hyperinsulinism is associated with maternal diabetes and gene defects affecting regulation of insulin production [Kapoor et al., 2009], and should be suspected when severe hypoglycemia (<2.6 mmol/L) without ketonuria in a well-nourished infant persists beyond the first 2 postnatal hours despite initiation of enteral feeds. Hypoglycemia from increased substrate utilization occurs during hypoxia or ischemia due to a shift from aerobic to anaerobic metabolism, sometimes compounded by decreased hepatic gluconeogenesis and ketogenesis. Brain glucose consumption is dramatically increased by seizures from any cause, and may exceed the capacity of cerebral energy metabolism in brain regions compromised by a recent ischemic insult. Inborn errors of metabolism may cause hypoglycemia due to enzyme deficiencies in glycogenolysis or gluconeogenesis, or as a result of hepatocellular dysfunction. The neurologic manifestations in these cases represent combined effects of the hypoglycemia and the metabolic disorder. Table 20-4 lists inborn errors of metabolism commonly associated with neonatal hypoglycemia. Neonatal hypoglycemia may be associated with congenital endocrinopathies affecting the hypothalamic-pituitary-adrenal axis as a result of major brain malformations of the hypothalamic-pituitary region, congenital adrenal hypoplasia, or hypothyroidism.
Table 20-3 Causes of Neonatal Hypoglycemia
| Category | Specific Disorders |
|---|---|
| Disorders of adaptation to extrauterine life | Prematurity Intrauterine growth retardation |
| Hyperinsulinism | Infants of diabetic mothers Maternal isoimmune disease Genetic defects of insulin secretion ABCC8, ATP binding cassette C KCNJ11, potassium channel J11 GLUD1, glutamate dehydrogenase GCK, glucokinase HADH, OH–acyl-CoA dehydrogenase SLC16A1, solute carrier 16-1 HNF4A, hepatocyte nuclear factor 4 alpha Beckwith–Wiedemann syndrome Maternal isoimmune disease Congenital disorders of glycosylation |
| Elevated glucose consumption | Hypoxia Global ischemia Seizures |
| Acquired or transient hepatic dysfunction | Post hypoxic-ischemic hepatocellular injury Infectious hepatitis Any cause of liver failure |
| Primary endocrine disorders | Hypopituitarism due to brain malformation Congenital adrenal insufficiency Adrenal hemorrhage Hypothyroidism |
| Inborn errors of metabolism | See Table 20-4 |
Table 20-4 Inborn Errors of Metabolism in which Neonatal Hypoglycemia is a Common Symptom
| Category | Specific Disorders |
|---|---|
| Disorders of carbohydrate metabolism | Hereditary fructose intolerance Fructose 1,6-biphosphatase deficiency Glycogen storage diseases, esp. I and III |
| Fatty acid oxidation defects | Medium-chain and long-chain acyl-CoA dehydrogenase deficiencies Carnitine palmitoyl transferase II deficiency Mitochondrial trifunctional protein deficiency Malonyl-CoA decarboxylase deficiency (MCD) |
| Defects in ketone body synthesis | HMG CoA lyase deficiency |
| Gluconeogenesis defects | Holocarboxylase synthetase deficiency (multiple carboxylase deficiency) Pyruvate carboxylase deficiency |
| Disorders of branched chain amino acid catabolism and related organic acidurias | Maple syrup urine disease Propionic acidemia Methylmalonic acidemia Isovaleric acidemia Ethylmalonic aciduria 3-methyl-glutaconic aciduria 2-methyl-3-OHbutyryl-CoA dehydrogenase deficiency (MHBD) |
The clinical signs and symptoms of hypoglycemia in the neonate are variable and nonspecific, exemplified by infants with hyperinsulinism. Signs and symptoms in the early stages may be dominated by signs of elevated circulating catecholamines: tachycardia, pallor, diaphoresis, irritability, jitteriness or tremor, and exaggerated tendon reflexes and neonatal reflexes (e.g., Moro reflex). Progressive depression of consciousness occurs as cerebral glucose stores are exhausted, associated with hypotonia, depressed reflexes, hypothermia, and often seizures. At its worst, uncorrected hypoglycemia at levels <1.0 μM in the absence of circulating ketone bodies will progress to coma, characterized by isoelectric electroencephalogram (EEG), cerebral edema, flaccid unresponsiveness, apnea, loss of brainstem reflexes, and circulatory collapse. Mild and moderate degrees of symptoms are rapidly reversed with glucose administration, whereas severe encephalopathy reflecting irreversible neuronal injury is not. Hypoglycemic coma is associated with irreversible neuronal necrosis and lasting neurologic deficits, with a predilection for parietal-occipital cortex and periventricular white matter [Burns et al., 2008]. Long-term sequelae may include microcephaly, cortical visual impairment, combined motor and cognitive handicap, and, in some cases, epilepsy [Tam et al., 2008].
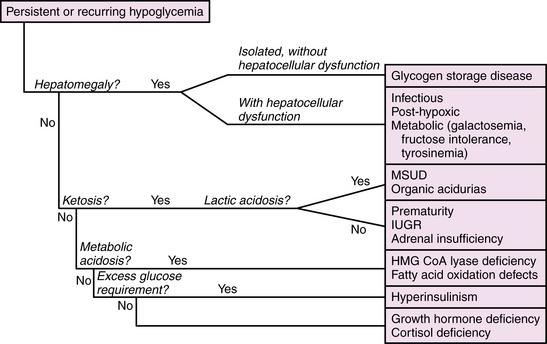
The management of neonatal hypoglycemia remains a controversial subject [Hay et al., 2009]. High-risk infants should be screened for hypoglycemia, and provided exogenous feeds, intravenous or enteral, at rates and concentrations sufficient to maintain adequate blood glucose concentrations. Bedside screening for low blood glucose should be performed in any infant who develops signs and symptoms of acute encephalopathy. Symptomatic infants should be treated while the first-stage evaluation proceeds, as outlined in Table 20-2. If ketones are absent and the exogenous glucose requirement is high (>10 mg/kg/min), then hyperinsulinism should be suspected and further evaluated by measuring insulin levels. A paradigm for assessment of hypoglycemia in neonates is shown in Figure 20-2. According to the “operational” approach to the definition of hypoglycemia, treatment of acute symptomatic hypoglycemia should proceed immediately for any infant with a blood glucose <2.5 mmol/L (46 mg/dL), with a minibolus of 200 mg/kg using 10 percent dextrose infused over 2 minutes, followed by continuous infusion of 10 percent dextrose starting at 8 mg/kg/min and increasing as necessary to maintain stable blood glucose levels >2.6 mmol/L. Monitoring the effect of glucose administration on clinical symptoms, particularly the level of consciousness, can help clarify the neurologic significance of the hypoglycemia, and the need for evaluation for other causes of encephalopathy.
Disturbances of Sodium Balance
Hyponatremia
Hyponatremia occurs when sodium losses exceed sodium or water intake, or reabsorption exceeds water excretion, or both. Hyponatremia is common in sick neonates due to immature or damaged renal mechanisms of salt and water balance or to perturbations in the hypothalamic-pituitary mechanisms regulating volume and osmolar status. Hyponatremia during the first 1–2 weeks of life in sick neonates is most commonly due to excess arginine vasopressin (AVP) secretion in association with other diseases such as lung disease, intracranial hemorrhage, pain, and surgical procedures, and rarely is due to congenital adrenal insufficiency. Causes of hypo- or hypernatremia are summarized in Box 20-2.
Box 20-2 Causes of Hyponatremia and Hypernatremia in Neonates
Causes of Hyponatremia


































The management of hyponatremic encephalopathy in neonates depends on the cause, the chronicity, and the severity of symptoms. Subacute or chronic (days to weeks) hyponatremia of mild degree (>120 mEq/L) should be treated by determining and correcting the underlying cause. Controversy surrounds the treatment of acute severe hyponatremic encephalopathy, which is usually associated with serum sodium concentrations <120 mEq/L. In such cases, seizures are usually resistant to standard anticonvulsants, and the encephalopathy frequently depresses respiratory drive and airway protective reflexes, all of which constitute a degree of urgency in favor of rapid correction. Rapid correction of hyponatremic seizures in infants and young children can be accomplished without neurologic complications by the use of 3 percent saline [Sarnaik et al., 1991]. The fact remains, however, that very little is known about neurologic complications from rapid correction of acute symptomatic hyponatremia in premature infants and critically ill term neonates who are already at high risk for neurologic injury from other causes. Until better data are available in this population, treatment should proceed with caution.
Hypernatremia
Neurologic manifestations of hypernatremia arise from hypertonic dehydration of neurons and glia, and may be compounded by secondary ischemic injury from hyperviscosity, cerebral venous thrombosis, or intracranial hemorrhage. Mild symptoms accompany mild elevations of serum sodium, generally <160 mEq/L, while severe encephalopathy usually occurs with elevations >170 mEq/L. The causes of hypernatremia are the inverse of those causing hyponatremia: excess sodium intake relative to sodium losses, or excess water losses relative to water intake or reabsorption, or both (see Box 20-2). Excess sodium intake occurs with administration of hypertonic intravenous solutions or from hypertonic oral feedings. Negative water balance is the most common cause of hypernatremia, caused by excess losses of hypotonic body fluids, defective renal water absorption due to nephrogenic diabetes insipidus, deficient pituitary secretion of AVP, or cerebral salt wasting. Hypothalamic-pituitary insufficiency is caused by acquired brain insults, such as meningitis or hypoxic-ischemic encephalopathy, and by midline brain malformations. Management of hypernatremia begins with assessment and correction of intravascular volume deficits with normotonic solutions or plasma expanders in sufficient amounts to restore perfusion to normal levels and to maintain urinary output. The guiding principle in treatment is to prevent cerebral edema by correcting the hypernatremia over 48–72 hours, decreasing the serum sodium concentration at a rate not exceeding 10–15 mEq/L per 24 hours.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


